The Processes of Ocular Scar Formation
The Processes of Ocular Scar Formation
Scar formation results from a complex array of processes that occur during wound healing and is triggered by tissue damage. During the scarring process the damaged tissue accumulates extracellular matrix (ECM) deposited by migrating cells, which later differentiate into contractile myofibroblasts or transformed epithelial cells.1 Tissue damage initiates the wound healing cascade, which is followed by an inflammatory phase during which immune cells migrate to the site of damage, releasing pro-inflammatory factors (cytokines, growth factors and chemokines).2 Growth factors and cytokines stimulate cell migration and proliferation to the wound site. The final stage of scar formation involves the remodelling of the local wound ECM, followed by the deposition of new matrix proteins under the control of growth factors3 and, finally, contraction of the local tissue into a scar.
Remodelling of the local ECM is critical to scar formation, and occurs via the secretion of matrix metalloproteinase (MMP) enzymes under the influence of cytokines and growth factors. It has been demonstrated that the severity of ocular scarring is dependent on the levels of MMP produced, where overexpression often produces exaggerated scarring.4,5 MMP inhibition is one potential mechanism by which scarring may be controlled.6
Growth Factors and Their Importance to Ocular Scar Formation
Growth factors are soluble proteins that are actively involved in many processes, including ocular development7,8 and wound healing.9–11 Growth factors are produced by a wide variety of cell types (see Table 1) and can act via autocrine (effect on the same cell), juxtacrine (affects adjacent cells with direct contact, such as epithelial cells)and paracrine (cells in close proximity) mechanisms.10
For simplicity, the growth factors have been classified into six roups,12 namely the transforming growth factors (TGFs), plateletderived growth factors (PDGFs), epidermal growth factors (EGFs), fibroblast growth factors (FGFs), insulin-like growth factors (IGFs) and hepatocyte growth factors (HGFs). Of the aforementioned growth factors, arguably the most important, and the most influential to ocular wound healing, are the TGFs.
TGFs can be considered the most important of the growth factors not only because they are implicated in the myofibroblast transition of fibroblasts13,14 and retinal pigment epithelia (RPE);13–15 they are also responsible for increased collagen and fibronectin deposition by many different cell types. Five TGF isoforms have been identified, of which only TGF-β 1, 2 and 3 are found in humans. Low levels of TGFPeng β have been shown to enhance cellular production of PDGF,16 which promotes cell proliferation and migration and has been shown tohave an autocrine effect in RPE, promoting cell growth.17 Tumour necrosis factor-alpha (TNF-α) is believed to be inhibitory to PDGFinduced migration.18
Growth Factors and Scarring in the Cornea
Cytokines and growth factors are key to the maintenance of the corneal epithelium. Interactions between epithelial cells and stromal fibroblasts involve cell-surface molecules such as integrins and connexins, ECM or cytokines.19,20 These play an important role in the development, homeostasis and wound healing capabilities of the corneal limbal niche (the principal habitat of the stem cell population that sustains the corneal epithelial mass).21
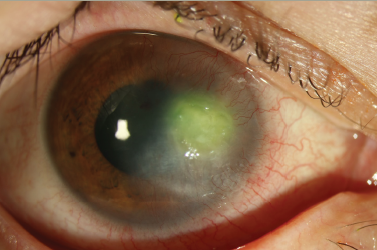
Deposition of fibrotic tissue and scarring can severely impair normal function of the cornea and clarity of vision (see Figure 1`). Both the opaque nature of the scar tissue and corneal contraction hamper the focusing of light and, as such, affect the vision of patients. The prevention of scarring is therefore crucial, and investigation of the implicating mechanisms can define the appropriate therapeutic strategies.
TGF-β isoforms, which are constantly expressed by the corneal epithelium,22,23 have been shown to regulate corneal epithelial, endothelial and fibroblast chemotaxis.24 TGF-β is a key player in scar tissue formation25,26 in the corneal stroma. Ordinarily, the basement membrane of the stroma prevents the diffusion of molecules (such as TGF-β); however, following injury or surgery, this barrier becomes compromised, allowing for diffusion.
TGF-β signalling also contributes to the recruitment of bonemarrow- derived cells to the wound site,27 which subsequently express fibronectin, one of the main ECM components of fibrous tissue.28 In addition, TGF-β induces α-smooth muscle actin (α-SMA)expression by the stromal fibroblasts, which makes them more contractile and is a typical feature of fibrosis.29,30 Of the three isoforms, TGF-β2 has been suggested to play the most vital part in scar formation during corneal wound healing, and its specific inhibition has reduced fibrosis.23 The TGF-βs act via the SMAD signalling pathways to onset α-SMA expression and actin stress fibres.31 SMAD proteins are involved in the signalling and inhibition of signalling of the TGF family. It is believed that fibronectin deposition by TGF-β stimulation is mediated by the c-Jun N-terminal kinase (JNK) pathway, a member of the mitogen-activated protein (MAP) kinase (MAPK) superfamily.32,33
Connective tissue growth factor (CTGF) is regulated downstream of TGF-β and, as such, fibroblast stimulation and ECM deposition by TGF-β may be under the control of CTGF.34 CTGF is essential (but not the sole requirement) for the TGF-β1-mediated transdifferentiation of fibroblasts to myofibroblasts following mechanical stress.35 Pharmaceutical intervention of corneal scarring can potentially focus on the inhibition of CTGF, as opposed to TGF-β, which has more physiological actions than CTGF in healthy tissue.36 TGF-β, the key player in corneal scarring, is still the most prominent pharmaceutical target as effective neutralisation on the wound site could reduce scarring after surgery or injury;37 this is especially the case as many of the downstream wound healing processes are under the influence of the TGF-βs.
Scarring of the Lens Capsule and the Role of Growth Factors
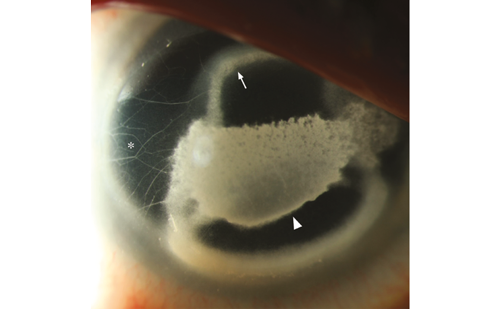
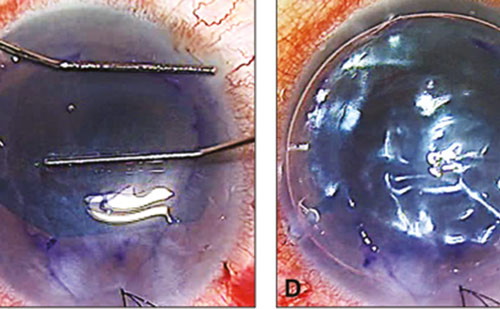
Cataract surgery is the most common eye operation worldwide. The most common complication is posterior capsule opacification (PCO), which can occur in to 20–40% of patients undergoing surgery34,35 (see Figure 2). As with other fibrotic disorders, TGF-β is highly implicated in PCO development.
The lens is an invagination of the surface ectoderm that is formed during embryogenesis.38–40 Loss of vision attributed to scar formation in the lens has been attributed to TGF-βs.19,20 As with other tissues, TGF-β upregulates the expression of α-SMA, promoting the epithelialto- myofibroblast transition of lens epithelial cells (LECs). Multiple isoforms of TGF-β have been shown to become deposited on matrix deposits that form on implanted intraocular lenses.41 Downstream mediators of the TGF-β2 pathway, including CTGF and α-SMA, were also found on capsular bag tissue analysed post mortem,42 suggesting LEC–myofibroblast transdifferentiation and matrix contraction.14,23
TGF-β is known to be present within aqueous humour,43 but is largely inactive as it is secreted bound to the propeptide latency-associated protein (LAP).44,45 Annes et al.45 suggested that the TGF-β–LAP complex,active TGF-β and TGF-β-binding protein have separate roles in wound repair.45 The latent TGF-βs, for example, are defined as ‘detectors’ that detect changes in the ECM or inflammation. When these ECM changes occur, the latent TGF-β form becomes activated.
Following cataract surgery, TGF-β levels in the eye rise,27,28 decreasing rapidly a few days after the surgery.46 Wormstone et al. demonstrated that contraction of a capsular (lens) bag after exposure to TGF-β2 for just two days was no different from that of a capsular lens bag exposed to TGF-β2 for the full 28 days.47 These data suggest that TGF-β initiated a cascade of events (presumably involving CTGF) leading to LEC transdifferentiation and scar formation.
As a target for pharmaceutical intervention, antibodies to TGF-β2 may be useful in the prevention of fibrosis and the downstream activation of CTGF.29,31 While monoclonal antibodies were successful in both in vitro and ex vivo PCO models, a rodent model of PCO failed to show any significant beneficial effect of the blocking antibody.48 No change in α-SMA staining was demonstrated (no prevention of LEC transition), although a decrease in capsular wrinkling was observed. This may demonstrate the need for more frequent administration of antibody, the prolonged release of antibody or the use of polyclonal antibodies to different downstream mediators of TGF-βs.
FGF has been shown to improve the survival of LECs after treatment with TGF-β2,49 as well as to promote survival of LECs in capsular bags from donor tissue.42,50 Basic FGF (bFGF or FGF-2) has also been implicated within PCO formation through activation by MMP-2 in the lens capsule.51 FGF-2 has been shown to inhibit TGF-β-induced apoptosis,49 as well as to actively promote the proliferation of LECs in vitro.52
Unlike FGF, EGF cannot overcome TGF-β-induced cell apoptosis.49 It is believed to promote the proliferation of cells in an in vitro posterior bag explant.53 The effect of EGF can be inhibited by blocking EGF receptors using a receptor antagonist. HGF has also been shown to induce LEC proliferation; this effect can be blocked using aneutralising antibody against the HGF receptor.54
The Influence of Growth Factors on Post-glaucoma Filtration Surgery Scarring
Glaucoma filtration surgery (GFS) is aimed at reducing the intraocular pressure (IOP) in patients that are non-responsive to conventional therapies for reducing elevated levels of IOP. The main cause of GFS failure is the conjunctival scarring response (see Figure 3).55 The use of mitomicin C (MMC) and 5-fluorouracil (5-FU) to prevent scarring has improved surgical outcomes following GFS.56 Unfortunately, these agents are associated with many potentially blinding side effects.57
The site of the wound healing response in GFS has been shown to contain a large number of growth factors.58 Much work has been undertaken to study how the modulation of these growth factors may influence the response of conjunctival fibroblasts and allow for more effective control of GFS scarring.
TGF-β is the key stimulatory growth factor in the aqueous fluid.59 As with other ocular tissues, TGF-β controls wound healing by stimulating fibroblasts to deposit ECM, promoting cellularproliferation and inducing the expression of MMPs.59,60
In a clinically validated in vivo model of GFS, a monoclonal antibody against TGF-β2 improved the surgical outcome with minimal destruction of local tissue.61 Unfortunately, subsequent human clinical rials did not show a similar significant effect.62 As with the rodent model of PCO,66 the failure of this clinical trial was attributed to the local ocular bioavailability of the antibodies, making it extremely difficult to obtain therapeutic antibody levels at the site of wound healing.63
Other promising therapeutic strategies targeting TGF-β in experimental GFS models include Tranilast ((N-(3u,4u)-dimethoxycinnamoyl) anthranilic acid) and suramin.64 Gene silencing has also shown promise for preventing GFS scarring, where short interfering RNA (siRNA) was used to downregulate TGF-β receptor II protein expression; the resultsdemonstrated reduced inflammation and deposition of ECM by fibroblasts in the conjunctiva.65
The contraction part of conjunctival scarring has been attributed to the action of TGF-β on fibroblasts, where it is believed to act via the Rho-associated kinase (ROCK) signalling network. Inhibitors of ROCK have been shown to reduce both cell tension and α-SMA expression in fibroblasts, thus reducing the ability of fibroblasts to transform into myofibroblasts and contract collagen.66 As such, ROCK provides an interesting target for pharmaceutical intervention of glaucoma scarring as, by inhibiting ROCK, the TGF-β-mediated scar formation may be inhibited.
The downstream activities of TGF-β in glaucoma, as with ot her wound healing situations, are influenced by CTGF. CTGF has been shown to be necessary for TGF-β stimulation of myofibroblast differentiation and collagen contraction;67 therefore, as with CTGF in the cornea, treatments may be possible to inhibit this growth factor, thus limiting the scarring potential of fibroblasts directly.68
The potential involvement of p38MAPK via its effects on CTGF and of the monocyte chemoattractant protein (MCP-1) has been studied.66 Therefore, the inhibition of p38MAPK has also been proposed as a possible future therapeutic option.
PDGF may also play an important role in post-GFS scarring. It stimulates fibroblasts, making them proliferate, migrate and produce ECM molecules, causing contraction, fibrosis and development of fibrotic membranes such as those observed in proliferative vitreoretinopathy. Agents capable of blocking PDGFBB have been tested in animal models of proliferative vitreoretinopathy (PVR) and may also be useful in glaucoma surgery.
Related to the TGF-βs are the bone morphogenic proteins (BMPs); these growth factors play an important role not only in embryonic development but also in cell proliferation, differentiation, apoptosis, angiogenesis and other biological functions.64,65 Increased expressionof BMP-6 mRNA and protein levels have been observed in scarred conjunctiva. The inhibition of BMPs could be a future therapeutic option for the control of scarring after GFS.69
Growth Factors in Proliferative Vitreoretinopathy
PVR is a major complication that occurs in approximately 10% of patients presenting with retinal detachment (RD). Retinal detachment is a clinical emergency where the retina becomes detached from the RPE layer. RD may result from physical trauma to the eye resulting in tractional pull of the retina. The fibrous scar (epiretinal membrane) that forms during PVR development dramatically increases the chance of failure of reattachment surgery due to the stiffness of the membrane. The effect of the epiretinal membrane (EM) formation is dependent on the site of scarring. EM formation proximal to or at the site of the macular can lead to vision loss and visual distortion and monocular dipilopia (see Figure 4).70 The migration and proliferation of many cell types, including RPE, fibroblasts and Müller glia, have been attributed to PVR development;33,69–72 RPE cells are believed to dedifferentiate and adopt a fibroblastic phenotype during PVR development.71
Where RPE migration is concerned, a tear in the retina or rhegmatogenous detachment allows growth-factor- and cytokinerich72–75 vitreous fluid to come into contact with the RPE layer and RPE cells, promoting their proliferation76 and migration77 into the vitreous and onto the retina where they form scar tissue (EM) on both sides of the retina. In a cat model of PVR, proliferation of RPE was shown as early as 24 hours after retinal detachment.78 During retinal scaring, both growth factors and cytokines have been shown to influence the expression of MMPs by RPE; in vitro RPEs express MMPs 1, 2, 3 and 9.79 In addition, retinal membranes from PVR patients have been shown to have elevated levels of those MMPs.80
The vitreous of patients with PVR has been shown to be high in both MMP-2 and MMP-9.4 Neutralisation of a combination of MMPs using antibodies was shown to be required to inhibit RPE-mediated gel contraction; individual MMP antibodies were not effective.71 A broadspectrum MMP inhibitor (ilomastat) was shown to inhibit collagen gel contraction by RPEs more effectively than the combined antibodies. The effects of ilomastat were shown to be reversible after replacement of the media, suggesting effective, reversible inhibition of contraction. MMPs are, as such, a valuable target for PVR development. It may be feasible to inhibit both the proliferation of RPE and MMP activity using a combination of a broad-spectrum MMP inhibitor plus an anti-inflammatory.71
Aside from the importance of cytokines such as TNF-α, which is believed to control intergrin expression and promote migration of RPE,77 growth factors play an important role in the formation of PVR.It has been suggested that HGF is important in the early stages of PVR development.81 This was realised based on the strong cellular staining of HGF on PVR specimens and relatively weak staining on scar tissue. Hinton et al. suggested that HGF, which acts mainly on epithelial cells, promotes cell division and migration of RPE in PVR. It has been hinted that HGF acts via an autocrine system in RPEs as they express both HGF and its receptors.82 HGF is suggested to promote the breakdown of RPE tight junctions,83 possibly as a result of downregulation of zona occuldens 1 (zo-1, a tight junction protein that is involved in cell–cell signalling).
TGF-β probably plays the most important role in the scaring response of the retina. Of the three TGF-βs expressed in mammals, TGF-β2 is the most important isoform for retinal scarring; indeed, a significant correlation exists between the levels of TGF-β2 expression and theseverity of retinal scarring.84 TGF-β2 mediates RPE production of collagen I and fibronectin,85,86 and promotes cell migration86 and the transition of RPE into the fibroblastic phenotype.87 CTGF again promotes the downstream activities of TGF-β,88,89 including RPE proliferation and matrix deposition.89 It is likely that the N-terminal of CTGF is responsible for the fibroblastic transition of RPE.89 Smad3 has also been implicated in the TGF-β-mediated dedifferentiation of RPE into fibroblasts,90 as Smad3-deficient mice failed to develop retinal scarring following detachment: it has been postulated that through Smad3, TGF-β2 (or possibly activin) induces the dedifferentiation of RPE into fibroblasts using TGF-β1, CTGF and PDGF as downstream mediators.90 It is likely that release of the basic dimer of PDGF (PDGF BB) by TGF-β is triggered via the Smad3 pathway. PDGFBB in turn promotes proliferation of RPE via its autocrine function,90 and this may be particularly important as TGF-β has a negative effect on the proliferation of RPE cells.91
While vascular endothelial growth factor (VEGF) is present in the vitreous of patients with PVR, it is not believed to play a major role in the scarring process. TGF-β has been shown to induce (via paracrine signalling) VEGF expression.92 VEGF is an important growth factor that is involved in angiogenesis,93–95 neovascularisation96 and diseases including age-related macular degeneration (AMD),97,98 retinopathy of prematurity99 and diabetic retinopathy.100 Hypoxia of the retina, either as a result of a change in the oxygen saturation in premature infants101,102 or as a result of retinal ischaemia in diabetic retinopathy,100,103 stimulates the release of VEGF from cells, including Müller cells104 and astrocytes.105,105
Protective Role of Growth Factors and Neurotrophins
The role of the neurotrophins in scar formation should also be discussed as these are frequently overlooked in the discussion of growth factors. Brain-derived neurotrophic factor (BDNF), neurotrophin-4 (NT-4, sometimes referred to as NT-4/5), nerve growth factor (NGF) and NT-3 are prevalent in the retina.107,108
Müller cells are a major source of BDNF109–111 and have been shown to promote ganglion cell survival through the use of Müller-cellconditioned media.110 BDNF is of particular interest as it has been shown to aid recovery of the retina following retinal detachment, not bypreventing cell death but by modulating the proliferation of Müller cells and decreasing their expression of GFAP.112 A later study demonstrated how both BDNF and bFGF2 may prevent ganglion cell apoptosis in a rat model of retinal detachment.113 By preventing ganglion cell apoptosis, neurotrophins may delay the inflammatory response following retinal detachment, thus delaying the progression of PVR.
Conclusions
This article covers the role of growth factors and fibrosis in some of the most clinically important and interesting situations in the eye including retinal scarring, lens capsule and post-GFS scarring. Scarring plays a major role in surgical failure and blindness in most countries of the world today. Fibroblasts, RPE, LECs and Müller cells play a major role in the formation of the scar tissue. Each step of the wound healing process that leads to scar tissue formation is under the control of inflammatory cytokines and growth factors. TGF-βs play a major role in every step of the wound healing process in the eye, by promoting the secretion of growth factors involved in cellular migration, proliferation, ECM deposition and myofibroblast formation. As such TGF-βs are still a key potential target for therapeutic intervention in the scarring process. Successful pharmaceutical intervention of scar tissue formation in the eye willrequire the innovative design of dosage forms to allow the optimal delivery of therapeutic agents at efficacious concentrations. It may be advantageous to deliver a combination of agents to the eye to inhibit different aspects of the scarring process. These agents may be anti-inflammatory to prevent the action of inflammatory cytokines and growth factors, or they may act by inhibiting the migration or proliferation of cells such as RPE; more importantly they may inhibit MMPs and thus prevent the remodelling of the ECM, which facilitates cell migration and ultimate scar tissue formation. There continues to be a major unmet clinical need for the control of ocular scarring.