Inflammatory myofibroblastic tumors (IMTs) are rarely reported, benign tumors. They are uncommon in the ocular structures. A four-year-old girl presented to us with bilateral proptosis. Subsequent radiologic and histopathologic investigations proved it to be bilateral IMT. This case report is being presented because of its rarity and some of the unique features of the condition seen in our patient.
Case Report
A four-year-old, apparently healthy girl presented to Queen Elizabeth Hospital, Kota Kinabalu two years ago, with a gradually progressive proptosis, lagophthalmos, and watering from her right eye for the past eight months, as noted by her mother. There was no blurring of vision, diplopia, pain, or redness noted. There was no history of trauma, nor symptoms suggestive of malignancy, thyroid abnormalities, connective tissue disease, or infection. She was well and active at home. There was also no family history of malignancy reported.
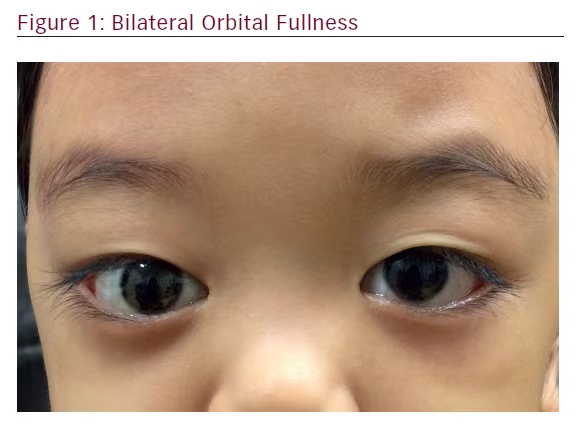
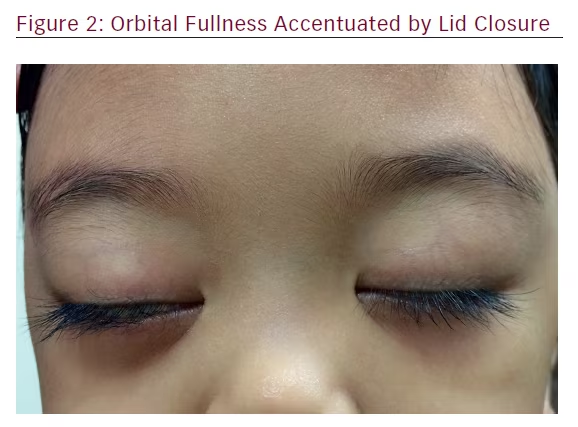
Examination of the child revealed an active girl with no features of dysmorphism. The visual acuities were 6/6 in both eyes. The conjunctivae in both eyes were mildly injected. The rest of the anterior, as well as posterior, segments were normal bilaterally. She was noted to have bilateral non-axial proptosis, which was worse on the right eye. The right eye was deviated downwards and inwards. Exophthalmometer measurements at 95 mm were 18 mm on the right eye and 16 mm on the left eye. No lagophthalmos or exposure keratopathy were present. Her extraocular movements were normal. There was no relative afferent pupillary defect noted (Figures 1 and 2).
Blood investigations consisting of full blood count, liver function tests, peripheral blood film, thyroid function, and work-up for sarcoidosis were all normal. Computerized tomography (CT) scan of the thorax, abdomen, and pelvis showed cervical lymphadenopathy. An ultrasound abdomen was also performed, but no metastatic lesions were noted.
Magnetic resonance imaging (MRI) of the brain and orbit revealed an extraconal lesion in the right supero-lateral aspect of the orbit measuring 1.2 x 1.36 x 3.4 cm. The lesion was isotense with grey matter in T1 and T2 weighted images. There was a homogenous enhancement post contrast injection. The mass was extending to the orbital apex, causing widening of the inferior orbital fissure. Erosion of the greater wing of sphenoid on the same side was noted. The left extraconal lesion measured 1.1 x 1.1 x 4 cm. There was extension of the mass into the apex of the orbit and the anterior aspect of the cavernous sinus. Minimal erosion of the greater wing of sphenoid bone was noted. However, no abnormal enhancements of the optic nerves were seen. There was enhancing of the dura along the superior sagittal sinus, which showed a normal signal void. The choroid plexuses on both sides were also enhancing on contrast injection. Based on the MRI report, the differential diagnoses were: granulocytic sarcoma, lymphoma, or neuroblastoma (Figure 3).
Subsequently, bone marrow aspirations (BMA) and lumbar puncture (LP) were performed by the pediatric hematology oncology team. BMA/ immunophenotyping were not suggestive of leukemia. Cerebrospinal fluid cytology also did not show any malignant cells. A CT scan revealed cervical lymphadenopathy, but no metastasis in the lungs or bones.
A month after first presentation, a right lateral supra-orbital keyhole craniotomy and excision biopsy of the lesions was performed. The highpower examination (HPE) of the lesion showed bland spindle-shaped cells arranged in interlacing fascicles and whorls with hyaline change in the stroma. No mitotic figures, cellular atypia, or granulomas were seen. The spindle cells were positive for vimentin and smooth muscle actin (SMA). However, a large part of the lesion was negative for desmin, caldesmin, glial fibrillary acidic protein (GFAP), cluster of differentiation 68 (CD68), von Willerbrand factor, cluster of differentiation (CD34), epithelial membrane antigen (EMA) or pan Keratin. A small fragment with inflammatory cells showed positivity for S100, vimentin, and CD68 (focal positive). However, it was negative for CD1a. These features were suggestive of IMT.
Postoperatively, the patient was started on oral prednisolone at a dose of 1 mg/kg once daily (QD) for six weeks. MRI of the brain and orbit was repeated three months postoperatively. The extraconal lesions in both orbits remained the same. The proptosis in both eyes also remained static. On a follow-up visit, nearly eight months post-operatively, fundus examination showed bilateral hyperemic, swollen optic discs with illdefined margins. An urgent MRI of the brain and orbit was ordered to rule out any mass effect/compression from the orbital IMT. The MRI reported extraconal lesions persisting in both the intraorbital regions. There was intense contrast enhancement with nodular enhancing choroidal plexus and dura in the anterior temporal region and sagittal sinus. Again, there appeared to be no significant reduction in the size of the lesions compared with the MRI, which was performed three months previously.
In view of the consistent MRI reports, the patient was referred to the pediatric hematology oncology unit. There, the patient was given intravenous (IV) methylprednisolone in a dose of 15 mg twice daily (BD) for a week and continued with prednisolone 15 mg BD for two weeks in a tapering dose. A month later, another follow-up with us showed a slight improvement in the papilloedema.
As the patient showed some improvement with the high-dose steroid, the patient was admitted by the pediatric hematology oncology unit. She was again given IV methylprednisolone 17 mg QD and continued with oral prednisolone, which was tapered off over a period of three months.
During a follow-up period of one year, the patient had three episodes of flare-up of the disease. The mother noted worsening of the proptosis. She was subsequently given IV methylprednisolone 17 mg BD for a week and a tapering dose of oral steroids for another five weeks. There was a significant improvement in the condition after the last dose. The latest MRI brain and orbit done six months previously revealed a stable disease with a mild right proptosis. The visual acuity of the patient remains 6/6 in both eyes and no signs of optic nerve damage were found.
Discussion
IMT is a rare pseudoneoplastic inflammatory condition. IMT is also referred to as pseudo-sarcomatous myofibroblastic proliferation, pseudotumor, and plasma cell granuloma.2 The World Health Organization has defined IMT as “a tumor composed of differentiated myofibroblastic spindle cells usually accompanied by numerous plasma cells and/or lymphocytes.”1–5 It commonly occurs in children and young adults, with no sexual or racial predilection.6,7 Due to its prevalence in pediatric patients, a differential
diagnosis of rhabdomyosarcoma should be kept in mind.8 IMT is a distinct mesenchymal neoplasm that arises in a variety of organs. Uncommonly, it may affect the central nervous system and rarely, the orbit. IMT was originally described in lungs, and other sites such as the mesentery, retroperitoneum and genitourinary tract.8 A benign condition, it is of unknown etiology and diverse morphology. The condition has the potential for local growth and recurrence. Rarely, there may be a low grade malignant transformation into sarcomas. The overall prognosis, however, is favorable.9,11,15
Orbital involvement with IMT, especially bilateral, as seen in our patient, is extremely rare. It may present as extraocular muscle enlargement, as well as intraconal or extraconal masses. However, a literature search showed this case to be the first to report bilateral orbital involvement.
The pathogenesis of IMT is thought to be idiopathic. Some possible causes include: surgery, trauma, T and B cell lymphoma, and autoimmune reaction. There is also a tenuous relationship with vasculitis, inferior vena caval thrombosis, and infections with mycobacterium avium–intracellulare complex, Epstein-Barr Virus, actinomycetes, and mycoplasma.5,11
MRI studies of our patient showed low signal intensity on T1- weighted images and high signal intensity on T2-weighted images,
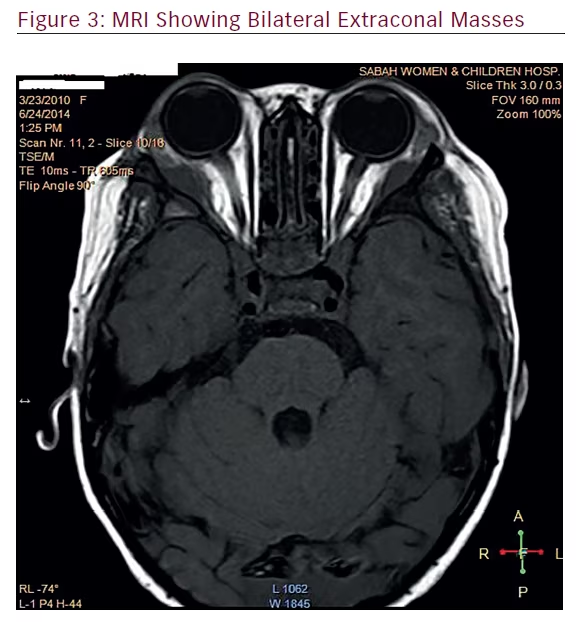
which correlated with several imaging studies on IMT. In our case, the MRI revealed bilateral extra-conal masses with extra ocular muscle infiltration. There were adjacent bony erosions, extending into the orbital apex and cavernous sinus bilaterally. Bone erosion was not seen in any of the reported cases so far. Only one other case has been reported to have bone invasion, which was reported by Lauwers et al. Imaging in that patient showed a left sino-orbital tumor with intracranial extension through the superior orbital fissure with bone erosion and sclerotic bone reaction.12
A biopsy of the orbital lesions in our case revealed proliferating spindle cells, which were suggestive of IMT. Immunohistochemistry for anaplastic lymphoma kinase-1 (ALK-1)1 is a useful indicator of chromosomal abnormality seen in IMT. However, ALK immunoreactivity has been reported only in 36% to 60% of cases.13 This expression was not seen in our patient. In HPE of our patient and in other case reports, the prominent cell type has been spindle cells.3 This tumor also invariably shows positive immunoreactivity for SMA, vimentin, and calponin.2,3,7,14
Surgical excision of the tumor is the treatment of choice of IMT. In orbital tumors it may not be possible to completely excise the tumor.5 Complete resection leads to cure and good prognosis. However, 25–35% of cases do recur. Any local recurrence or aggressive behavior of IMT is attributable to an incomplete resection of the mass.3,7,15
In the present case, it was determined that surgical excision would prove to be too risky to attempt. In such non-resectable cases corticosteroid therapy and chemotherapy have been suggested as a good alternative. Our patient also showed significant improvement after multiple sessions of IV methyprednisolone injections.2–4,12 Radiotherapy is used for recurrence or unresectable tumors. Usually high doses are required which may prove a limiting factor.3
In conclusion, IMT is a rare pseudo-neoplastic inflammatory tumor, which is difficult to diagnose. Involvement of the orbit, especially bilateral, is unusual in this condition. Bony erosion of the orbit is also a rare feature which is seen in this case. This case highlights that high dose corticosteroids are a viable option in those patients in whom surgical resection is not possible.