Lysosomal storage disorders (LSDs) are a group of more than 50 inheritable disorders.1,2 Although individually rare, they collectively affect approximately one in 5,000 live births.3 Defective metabolism of proteins, carbohydrates or lipids resulting from deficiency of one of the many lysosomal enzymes leads to pathological accumulation of substances within the lysosomes. This lysosomal accumulation triggers an insidious cascade of processes generally leading to progressive tissue damage and organ failure.
Lysosomal storage disorders (LSDs) are a group of more than 50 inheritable disorders.1,2 Although individually rare, they collectively affect approximately one in 5,000 live births.3 Defective metabolism of proteins, carbohydrates or lipids resulting from deficiency of one of the many lysosomal enzymes leads to pathological accumulation of substances within the lysosomes. This lysosomal accumulation triggers an insidious cascade of processes generally leading to progressive tissue damage and organ failure.
Most physicians have difficulty recognising patients in an early stage of the disease, as each patient is unique in terms of age of presentation, cluster of presenting symptoms, rate of disease progression and co-morbidities. Symptoms may initially seem harmless and may be suspected to be due to another, more common disease.4 Hence, valuable time may elapse before the patient is diagnosed and considered for therapeutic intervention with enzyme-replacement therapy (ERT), if available for that particular disorder.5
With age, progression of the disease generally has a strong impact on the health and quality of life of affected patients, and life-threatening complications significantly reduce life expectancy.1,5
The early signs and symptoms of LSDs can include pathognomonic ophthalmic features.6–8 It is thus imperative that ophthalmologists are familiar with these signs, even if they will encounter such an LSD patient maybe only once during their ophthalmic career. In this article, we will describe the disease characteristics and ocular features of five LSDs that are currently treatable with ERT: Fabry disease, mucopolysaccharidosis (MPS) types I, II and VI and Gaucher disease.
Fabry Disease
Fabry disease is a life-threatening LSD characterised by a deficient activity of the lysosomal enzyme α-galactosidase A.9 This deficiency results in a diminished ability to catabolise certain glycolipids. Progressive lysosomal accumulation of undegraded globotriaosylceramide (GL-3) occurs in many cell types throughout the body, e.g. in vascular endothelial cells, connective tissue, kidneys, heart, brain, nerves and cornea. The initial insult of substrate accumulation leads to secondary tissue damage and irreversible organ failure in a cascade of overlapping steps.10 Fabry disease follows an X-linked inheritance pattern; however, females who carry the defective gene can also develop variable clinical signs and symptoms partly based on skewed X-chromosome inactivation.11,12, The estimated prevalence of Fabry disease is 1:40,000 males.9
Clinical Course
In ‘classic’ Fabry disease, early symptoms typically arise in childhood or adolescence and generally include neuropathic pain, angiokeratoma, hypohidrosis, gastrointestinal disturbances, ocular manifestations and exercise intolerance. Neuropathic pain is often the earliest symptom in affected males and females. Two types of pain are common: acroparesthaesia (constant burning or tingling in the extremities) and acute, episodic pain attacks originating from the hands and feet triggered by illness, exercise, fever, stress or weather changes. As the disease progresses, life expectancy is substantially decreased by renal dysfunction, cardiac complications and cerebral involvement.9,13 Kidney involvement manifests as albuminuria/proteinuria and impaired glomerular filtration rate as early as in the second decade of life, and patients typically progress to end-stage renal disease in their 40s. Cardiac complications include left ventricular hypertrophy, arrhythmias, valvular abnormalities, myocardial infarction and cardiac failure. Central nervous system complications may include transient ischaemic attacks and early stroke. In heterozygous females, the disease spectrum spans from absence of symptoms to classic Fabry disease with severe renal, cardiac and/or neurological complications. As the initial Fabry symptoms can mimic those of more common disorders, many patients are not diagnosed until adulthood and, on average, there is a delay of a decade or more between symptom onset and diagnosis.14,15
Ocular Features
Cornea
Distinctive corneal opacities play an important role in the early detection and diagnosis of Fabry disease as they are reported to be present almost universally among male Fabry patients and in a high proportion (>70%) of heterozygous females.16,17 In some cases they cause light sensitivity.18,19 In the absence of retinal vascular occlusion and ischaemic optic neuropathy, visual acuity is typically not impaired. In the early stages, the opacities appear as a fine haze that is mostly diffuse and involves the whole cornea.20,21
Gradually, pale grey to brownish/yellowish spiral streaks will form in a vortex pattern in the corneal epithelium (‘cornea verticillata’), extending from just below the centre of the cornea to the periphery (see Figures 1a and 1b). Cornea verticillata is best observed by slit-lamp examination using a wide-slit beam angled from the side and utilising the light that is backscattered from the iris in order to evaluate the fine opacities. Cornea verticillata has been reported in ~95% of affected males and in ~70% of affected females, in whom it can even be the only apparent manifestation of the disease.20 The exact mechanism of formation of cornea verticillata is still unknown, but it has been postulated that formation of subepithelial ridges resulting from GL-3 deposition in the sub-epithelial area of the cornea causes variations in density on ophthalmological examination. Besides Fabry disease, cornea verticillata may be observed in patients being treated with a variety of drugs including amiodarone, chloroquine, amodiaquine, meperidine, indomethacin, chlorpromazine and tamoxifen.
Retina
Retinal vascular changes in the form of narrowing of arterioles and corkscrew-like exaggerated dilatation (tortuosity) of retinal and conjunctival veins are not uncommon, particularly in males (see Figure 2). These vessel abnormalities are most prominent in the posterior pole.19,20 In addition, hypertensive retinopathy or retinal oedema can develop as a result of vascular and renal Fabry disease pathology.
Conjunctiva
Conjunctival vessels with a corkscrew-like appearance and aneurysmal dilatations are frequently observed (see Figure 3).
Lens
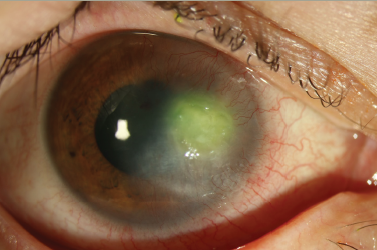
Anterior capsular or subcapsular deposits in the lens have been described. Classically, whitish, granular spoke-like deposits in the posterior lens constitute the ‘Fabry cataract’.18,19
Vision
Ocular lesions rarely affect vision; however, central retinal artery occlusion causing sudden visual loss has been described, as have cases of ischaemic optic neuropathy.19,22–24
Lacrimal Secretion
Reduced lacrimal secretion may occur due to vascular involvement or GL-3 deposition in the lacrimal glands.25
Enzyme-replacement Therapy
Recombinant human α-galactosidase A (Fabrazyme®, agalsidase beta) appears to offer the best choice in patients with Fabry disease based on evidence from controlled clinical trials and open-label studies.26–29 With agalsidase beta therapy, complete normalisation of storage of GL-3 in a variety of cell types, stabilisation of kidney function in patients with relatively preserved kidney function and reduction of the probability of a major clinical event may be anticipated. Responses of ophthalmological pathology to ERT have not yet been studied in formal clinical trials.
Mucopolysaccharidosis I (Hurler, Hurler-Scheie and Scheie Disease)
MPS I is an autosomal recessive inborn error of metabolism caused by mutations in both alleles of the gene encoding the lysosomal enzyme α-L-iduronidase.30 It represents the most common disorder of the group of seven MPS, with an estimated prevalence of 1:100,000 newborns. The enzyme deficiency leads to the inability of lysosomes to break down the glycosaminoglycans (GAGs) dermatan sulphate and heparan sulphate, which are important components of the extracellular matrix surrounding cells and integral parts of the cell membranes. They play a major role in the structure of the cornea, connective tissue and cartilage, as well as in joint fluids, and their metabolic degradation is essential for normal growth and tissue homeostasis. Progressive accumulation of undegraded GAG occurs in virtually all tissues and gradually causes tissue and organ dysfunction, with eventual cellular death throughout the body.30–32
Clinical Course
The wide spectrum of clinical disease spans from the most severe form (Hurler disease) to less aggressive forms of the disease (the Hurler-Scheie and Scheie phenotypes).31–33 In Hurler patients, the disease progresses rapidly. Patients are commonly diagnosed at around the age of nine months and cognitive defects and developmental delay usually become apparent before the age of two years. Patients will be severely mentally retarded and have pronounced somatic disease manifestations at the time of early demise, often before the age of 10 years. In more attenuated cases, joint pain and stiffness with difficulty in using the hands (see Figure 4a), corneal clouding, ear–nose–throat infections and recurrent umbilical hernia generally manifest before the age of two years. Coarse facial features, hepatomegaly, hearing difficulties and delayed speech development are common. Life expectancy in attenuated forms may be limited to <20 years, whereas Scheie patients with fewer physical symptoms may survive well into adulthood (see Figure 4b). However, cardiac insufficiency or complications of, for example, spinal surgery may reduce life span if cause-specific treatment and adequate supportive care
are not administered.
Ocular Features
The main ocular manifestations of MPS I include corneal clouding, retinal pigmentary degeneration, glaucoma and optic nerve abnormalities.6 Prominent, wide-set eyes (shallow orbits, hypertelorism) may be seen in severely affected patients.
Cornea
Corneal opacities are universally present in all forms of MPS I.31 Fine grey punctate opacities are first seen in the anterior stroma; later, as the disease progresses, they are also seen in the posterior stroma. Progressive corneal opacification is typically diffuse and is associated with photosensitivity and reduced central visual acuity. There are reports of denser clouding in the peripheral cornea in MPS disease with good visual acuity as a result of minimal central corneal involvement, especially in attenuated cases.34 Corneal grafts may improve visual acuity in such cases (see Figure 5). In severe cases, vision is impaired in early childhood and nystagmus may be present. If corneal clouding is detected it is important that the ophthalmologist considers a possible diagnosis of MPS I, particularly in the presence of other suspect clinical symptoms. However, other diseases should also be considered in the differential diagnosis (see Table 1).
Excessive deposition of GAGs within stromal cells (keratocytes) and within the extracellular stromal matrix may account for loss of corneal transparency. Numerous massively involved keratocytes may give the cornea a hypercellular appearance on light microscopy, and electron microscopy may show intralysosomal vacuoles loaded with mucopolysaccharides. Most important are the alterations in collagen fibre architecture (increased fibril diameter, fibrous long-spacing collagen) and disorganisation of collagen fibril packing within the lamellae. Furthermore, a decrease in the metabolic activity of endothelial cells due to abnormal storage may contribute to the pathology due to deregulation of stromal hydration causing increased corneal rigidity.35 Other light/electron microscopic observations in the cornea can include lysosomal inclusions in the corneal epithelium and attenuation of the Bowman membrane.36
Retina
Pigmentary degeneration, initially affecting the outer retinal layers, is not uncommon and photoreceptor loss may result in initial night blindness and subsequent decreased peripheral vision. The aetiology of retinal pigmentary degeneration is poorly understood, but it may be assumed that GAG accumulation is the underlying basis.
Glaucoma
Increased intraocular pressure is not uncommon in MPS I.31 Accumulation of incompletely degraded GAGs in the trabecular meshwork has been suggested as the primary cause of glaucoma in severe MPS I. Also, it was hypothesised that the meshwork is affected to a greater extent than other anterior segment structures, leading to open-angle glaucoma with progressive, permanent loss of vision and light hypersensitivity. In some patients, progressive shallowing of the anterior chamber and subsequent angle-closure glaucoma with sudden decrease of vision has been described.37
Optic Nerve
In severe MPS I, optic atrophy may develop after long-standing optic nerve head oedema, secondary to increased intracranial pressure and hydrocephalus. However, such patients may not live long enough to develop optic atrophy. Other causes of papilloedema that have been proposed include accumulation of GAGs in ganglion cells with subsequent axonal degeneration, compression of the optic nerve by thickening of meninges secondary to accumulation therein and compression of optic nerve axons due to scleral and septal thickening.38
Enzyme-replacement Therapy
Early diagnosis of MPS I opens up the possibility of ERT with recombinant human α-L-iduronidase (Aldurazyme®, laronidase). This can alleviate existing MPS I symptoms and would forestall the progression of the disease to irreversible and possibly life-threatening conditions.32 Stabilisation of ocular findings and visual acuity has been reported in five out of eight laronidasetreated MPS I patients during a four-year follow-up period.39
Mucopolysaccharidosis II (Hunter Disease)
MPS II is caused by a mutation in the gene that encodes the lysosomal enzyme iduronate-2-sulphatase.30 The disorder is characterised by an X-linked inheritance pattern, but females may also develop disease manifestations partly due to skewed X chromosome inactivation. Estimates of the prevalence ranging from 0.9:100,000 to 4.5:100,000 have been reported.40–42 Progressive deposition of undegraded GAGs (dermatan an heparan sulphate) occurs in various tissues and organs, but age at onset of symptoms and rate of disease progression are variable.30,42
Clinical Course
At the severe end of the spectrum, patients share many of the clinical features seen in MPS I Hurler patients. Patients are typically diagnosed between the ages of 18 and 36 months.43 Gradually, they lose acquired skills that may be equivalent to three to five years. Due to pronounced somatic involvement in conjunction with progressive neurodegeneration (mental retardation), patients generally die in adolescence. At the attenuated end, patients have a later onset of symptoms, no or minimal neurological dysfunction and milder, more chronic somatic involvement.31,42,43 The wide array of symptoms includes (among others) mild facial dysmorphism, exophthalmos, hypertelorism, short stature, communicating hydrocephalus, skeletal deformities, hepatosplenomegaly, recurrent inguinal hernia, joint stiffness and contractures, recurrent ear infections and progressive hearing loss. These patients may survive until their 50s or beyond, although they remain at risk of developing life-threatening respiratory and cardiac complications beyond their second decade of life.
Ocular Features
Cornea
Severely affected MPS II patients do not have corneal clouding and, therefore, this feature may be helpful in separating patients with MPS I from those with MPS II. More attenuated cases can have discrete corneal opacities that do not impair visual acuity and are detectable only by meticulous slit-lamp examination.
Retina
Pigmentary retinopathy has been described in patients with the severe form and, to a lesser degree, in the attenuated form.44–46 Apart from night blindness, visual impairment based on retinal degeneration is rare.43
Optic Nerve
Papilloedema in the absence of increased intracranial pressure with subsequent optic atrophy is not uncommon, and may result from scleral thickening due to GAG deposition.38,47
Glaucoma
Glaucoma is uncommon in MPS II.
Enzyme-replacement Therapy
Recombinant human iduronate-2-sulphatase (Elaprase®, idursulfase) was recently approved as a treatment of patients with Hunter disease based on evidence demonstrating its capacity to improve walking distance and joint mobility and to reduce hepatosplenomegaly.48 To date, there are no reports describing therapeutic responses in relation to ophthalmological features.
Mucopolysaccharidosis VI (Maroteaux-Lamy Disease)
MPS VI is an autosomal recessive disorder caused by mutations in both copies of the gene encoding the lysosomal enzyme N-acetylgalactosamine 4-sulphatase (arylsulfatase B).30 Dermatan sulphate, a key compound of connective tissue, progressively accumulates within lysosomes in a variety of tissues and organs including the skeleton, skin, tendons, heart valves, spleen, liver, airways and cornea. The pathological depositions trigger cellular dysfunction and death and result in progressive tissue and organ damage. Estimated prevalences range from 1:238,000 to 1:300,000.49
Clinical Course
As with other MPS, the clinical presentation in MPS VI varies widely: severe patients may present early in life with aggressive, debilitating disease manifestations, and patients at the other end of the spectrum show a more slowly progressive course.30,49 Patients with the severe form may seem normal at birth, but present in early childhood with an array of symptoms often resembling symptomatology seen in MPS I disease. However, mental retardation does not occur. Neurocognitive development may be delayed due to functional and visual impairments. In the first year of life, rapidly progressive skeletal deformities (dysostosis multiplex), hepatosplenomegaly, corneal clouding, coarse facial features, enlarged tongue and restricted joint movement due to contractures are noticed.
Growth almost stops at the age of six to eight years. Other complications include hearing loss, recurrent ear–nose–throat infections and hernias, obstructive airway disease/sleep apnoea, hydrocephalus, heart valve dysfunction, nerve entrapment syndromes and blindness. Severely affected patients die in the second or third decade of life particularly due to cardiopulmonary disease, infections or complications related to surgical procedures. Patients with a more attenuated form may survive until well into adulthood.
Ocular Features
Cornea
Moderate to severe corneal clouding is common, especially in cases with more attenuated disease,6 and frequently leads to visual impairment.
Retina
Although retinopathy has been reported in some cases,50,51 it appears to be a rare feature in MPS IV. Night blindness may be reported.49
Optic Nerve
Optic disc swelling is commonly observed and may progress to optic nerve atrophy due to a combination of increased intraocular pressure, GAG storage in optic nerve ganglion cells and/or compression effects.49
Glaucoma
Increased intraocular pressure is a common finding, but should be confirmed by combining applanation tonometry with pachymetry as corneal thickness may be increased. Glaucoma may be of the angle-closure or open-angle type (GAG deposition in the trabecular meshwork).52
Enzyme-replacement Therapy
Recombinant human arylsulfatase B (Naglazyme®, galsulfase) is available for the treatment of patients with Maroteaux-Lamy disease. Clinical studies have demonstrated that ERT is able to rapidly reduce urinary GAG excretion and improve endurance,53 but potential effects on the ophthalmological manifestations have not yet been reported.
Gaucher Disease
Within the group of LSDs, Gaucher disease is the most common enzyme deficiency disorder and is inherited in an autosomal recessive manner. The underlying cause is a deficiency of the enzyme acid β-glucocerebrosidase, which is normally involved in the degradation of complex glycosphingolipids.54 Progressive lysosomal accumulation of glucocerebroside occurs throughout the body in tissues of the monocyte–macrophage system, and symptoms arise due to cellular dysfunction and displacement of such normal cells by lipid-laden ‘Gaucher cells’. The prevalence of Gaucher disease in the general population is around 1:60,000, but may be as high as 1:1,000 among Ashkenazi Jews.54
Clinical Course
The spectrum of clinical onset and disease progression is remarkably heterogeneous and actually represents a disease continuum. Most patients suffer from the non-neuronopathic (type 1) form. In acute, rapidly fatal neuronopathic (type 2) or chronic neuronopathic Gaucher disease (type 3), patients also display neurological manifestations.55 Bone marrow infiltration with Gaucher cells, which causes displacement of haematopoietic elements, seems to be the first step in the aetiopathology of skeletal manifestations, which are common.56,57 Bone crises are more prevalent in children than in adults, but can occur for the first time in the third to seventh decade. Bone fractures are not uncommon. Other key manifestations include hepatosplenomegaly, easy bruising or bleeding (thrombocytopoenia), fatigue (anaemia), gastrointestinal complaints, pulmonary disease and growth retardation.55,56,58 If the disease is diagnosed early in life, the subsequent clinical course will, in general, be more severe.58 Unfortunately, even in such cases prolonged diagnostic delays occur that may result in even more severe disease manifestations.59 Chronic (type 3) neuronopathic disease includes several variants.60 Patients may exhibit prominent neurological abnormalities with relatively mild visceral involvement. A typical feature in these patients is the early development of an isolated horizontal supranuclear gaze palsy characterised by slowing of conjugate horizontal saccadic eye movements. Other patients may have subtle neurological abnormalities with severe systemic disease, or systemic manifestations in combination with cardiac valve calcifications, corneal clouding, oculomotor ataxia and mental retardation.
Ocular Features
Conjunctiva
Pingueculae, prominent yellowish lesions of the bulbar conjunctiva, may appear on both the nasal and temporal sides of the corneoscleral limbus and often remain unnoticed by the patients, although they may be more prevalent than in the general population (see Figure 6). They have been shown to contain lipid-engorged macrophages.61
Cornea
Milky-white opacities, presumably caused by abnormal lysosomal glucocerebroside deposits, have been reported to occur in the corneal epithelium (posterior stromal keratocytes), anterior chamber angle, ciliary body, pupillary margin, vitreous body and choroid.62,63 Most of the reported cases have been in neuronopathic disease.64–66
Retina
Clusters of Gaucher cells on the inner surface and within the inner layers of the retina have been reported (see Figure 7).66 Whether they are a sign of neuronal involvement with reduced electrophysiological responses is still under investigation. Evidence for cherry-red spots in the macula in Gaucher disease is questionable.64 Choroidal neovascularisation has been described in combination with extensive retinal pigment epithelial alterations.61
Saccade Initiation Failure
Patients with chronic neuronopathic type 3 disease typically develop saccade initiation failure characterised by ‘lock-up’ of the eyes at the limit of gaze due to a paucity of quick phases. It can be difficult to detect clinically, but is readily revealed as missed quick phases during induced optokinetic and vestibular nystagmus.60
Enzyme-replacement Therapy
Therapy with recombinant human β-glucocerebrosidase (Cerezyme®, imiglucerase) is very well tolerated, highly safe, efficacious in ameliorating symptoms and signs in most disease areas and effective in preventing further disease deterioration or development of irreversible manifestations.55,67–70 Splenectomy can be avoided, debilitating skeletal complications can be averted in many and the average life expectancy of Gaucher patients has increased, indicating a positive effect on reducing morbidity. Reported evidence of ophthalmological responses is limited, but reversal of loss of vision has been reported.71
Diagnosis and Genetic Counselling
In a patient with an LSD presenting with early symptoms and signs, it is likely that the consulted physician will first suspect a common, less serious disorder. However, regular follow-up visits will bring to light an increasing number of abnormalities, and may eventually raise the index of suspicion of a more serious disorder, requiring further diagnostic work-up.
Ophthalmological manifestations are frequent in MPS I, II and VI and Fabry disease and may even be the presenting signs or symptoms that lead to clinical diagnosis. Eye manifestations also occur in patients with Gaucher disease, particularly in the neuronopathic variant. If a clinical suspicion of LSD exists, the clinician should first request an enzyme activity assay. The most efficient and reliable method to confirm the diagnosis is an assay using peripheral blood leukocytes and an artificial substrate.
Although knowledge of the specific genetic mutation may be of some help in predicting the severity of the disease in a specific case, a more important role for genotyping relates to screening and genetic counselling of family members of the affected individual. Genetic testing of siblings, parents still at a reproductive age and members of an extended family in case of consanguinity is essential after the diagnosis has been established. The whole procedure should be accompanied by counselling by a qualified and knowledgeable genetic counsellor or geneticist, before and after testing, using validated techniques. Parents should be informed about the likelihood that siblings, relatives and future children will inherit the disease. Currently, pre-natal diagnosis can be established through enzyme deficiency testing using chorionic villi or cultured amniocytes, or by molecular analysis.
Screening of Large Populations
To identify undiagnosed cases with an LSD, large-scale screening programmes have been undertaken.72 In 2004, 296 German ophthalmologists participated in such a screening programme. Out of a total of 29,998 patients examined, 20 were identified for further testing on the basis of potential ophthalmic signs. Enzyme activity and genetic analyses were performed in these 20 patients, resulting in a confirmed diagnosis of Fabry disease in five patients and MPS I in one patient.
Ophthalmological Examination in Lysosomal Storage Disorders
Thorough ophthalmological evaluation should include assessment of best-corrected visual acuity, examination of the visual fields and assessment of strabismus. Corneal and lens opacities, as well as signs of optic nerve oedema or atrophy, can be detected by slit-lamp examination and direct or, preferably, indirect ophthalmoscopy. Optic disc changes may be masked by the corneal clouding, preventing proper visualisation of the retina and optic disc. Increased light sensitivity or photophobia will be evident during examination.
Posterior Fabry cataract is best observed by slit-lamp examination using retroillumination. Cornea verticillata is best observed by slit-lamp examination using a wide-slit beam angled from the side and utilising the light that is backscattered from the iris in order to exaluate the fine opacities. Fundus photographs are very useful in monitoring retinal pathology. Retinal dysfunction can be demonstrated by colour vision testing (e.g. Ishihara or Hardy-Rand-Rittler pseudo-isochromatic plates, Farnsworth Panel D-15 colour caps test) and full-field flash electroretinography (ERG). Retinal angiography may be helpful in cases with impaired perfusion. Visual evoked potentials may be used to objectively measure the cortical response to light.
Increased rigidity of the cornea and sclera may falsely lead to a diagnosis of glaucoma.36 Therefore, diagnosis of true glaucoma requires intraocular pressure measurement in combination with corneal pachymetry. Abnormalities of tear secretion may be demonstrated in patients with Fabry disease. In patients with neuronopathic Gaucher disease, eye movement disturbances should be evaluated on a regular basis. It is advised to take one or more facial, slit-lamp and fundus picture to document ocular and dysmorphic characteristics over time. Ocular status should be documented once the diagnosis of LSD is ascertained and should be monitored annually, particularly if ophthalmological pathology has been found.
Opthalmological Treatments
In addition to ERT to treat systemic disease manifestations, the ophthalmologist can provide specific ophthalmic care. Visual complaints should be treated to prevent blindness. Corneal grafts or phacoemulsification should be considered if marked opacification causes visual complaints, although risks of anaesthesia and prognosis are to be evaluated carefully. Re-accumulation of substrate may occur over time. Prescription of spectacles may be indicated. Lubricant eye drops can be prescribed for Fabry patients if tear production is deficient.
Conclusion
Ophthalmological manifestations are frequent in MPS I, II and VI, Fabry disease and neuronopathic Gaucher disease, and may even be among the presenting symptoms leading to clinical diagnosis. Because ERT is available, delay in diagnosis should be minimised and patients should be prevented from developing irreversible complications of these multiorgan diseases. The ophthalmologist plays a vital role here. Patients should be evaluated periodically by an ophthalmologist as part of the multidisciplinary disease management approach, irrespective of whether or not they are treated with ERT.