Scleritis refers to a heterogeneous group of diseases characterized by inflammation of the sclera, which may also involve the cornea, adjacent episclera, and underlying uveal tract.1 In contrast to episcleritis, scleritis is associated with significant ophthalmic and systemic morbidity.2 Patients who are not appropriately diagnosed and treated are at high risk of vision loss owing to the progressive destruction of the eye and other associated ophthalmic complications. Moreover, the presence of scleritis can be the initial manifestation of a potentially lethal systemic vasculitis or can be the sole sign of active systemic disease of an already diagnosed inflammatory disorder.3 Therefore, the recognition and prompt treatment of scleritis could not only protect the eye, but also prolong the life of the patient.
Specific etiologies of scleritis, varying from idiopathic to autoimmune to infectious, portend variable disease severity and outcome. Scleritis sometimes occurs in an isolated fashion, without evidence of inflammation in other organs. However, in up to 50% of patients, scleritis is associated with an underlying systemic illness such as rheumatoid arthritis (RA) or granulomatosis with polyangiitis (Wegener’s).1 Infection is an important but rare cause of the scleritis, occurring in about 5–10% of all cases.4 Geographical location should be considered, as infectious scleritis is more commonly seen in the southern area of the United States.5 However, owing to the similarity of its presentation, infectious scleritis is often initially managed as autoimmune, potentially worsening its outcome.
Clinical features
Scleritis can occur in any age group, but most commonly presents between the fourth and sixth decades of life; women are affected approximately twice as often as men.6,7 The primary clinical sign of scleritis is redness associated with severe pain. The redness has a bluish red appearance, tends to progress with time and can be sectorial or involve the whole eye. The pain is described as dull, aching, or boring and it may be severe and constant; it often awakens patients from sleep and is poorly responsive to analgesics. Patients complain of deep pain that radiates from the eye to the forehead, orbit and even the sinuses in some instances. It is exacerbated by touching the eye or by pressing the periocular area. Other complaints may include tearing, photophobia, and decreased vision (especially in posterior scleritis).2,7
Slit lamp examination shows edema of the episcleral and scleral tissue, with congestion of the deep episcleral plexus. The use of topical vasoconstrictors has minimal effect on these vessels and in contrast to episcleritis, the redness of scleritis will not be resolved with the instillation of 10% phenylephrine or 1:1000 epinephrine.
Scleritis is associated with a number of ophthalmic complications that can lead to loss of vision. These include keratitis, uveitis, glaucoma, exudative retinal detachment, and macular edema.2,7
Scleritis can be further classified into anterior and posterior forms using the Watson and Hayreh classification.2 This classification is useful in determining the severity of the inflammation and as a guide for treatment. Anterior scleritis is much more frequent and can be further divided into diffuse, nodular, necrotizing and necrotizing without inflammation (scleromalacia perforans).8
Scleritis subtypes
Anterior sclertitis
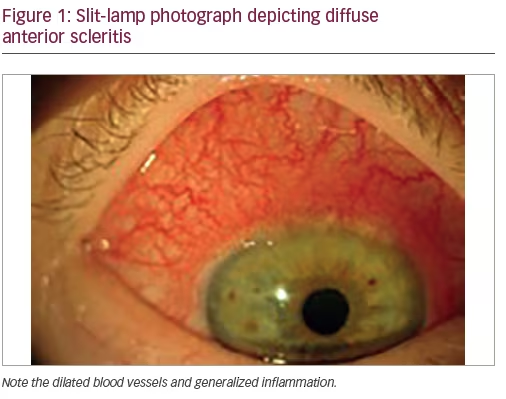
Diffuse anterior scleritis (see Figure 1)—the inflammation of diffuse scleritis is generalized, has an insidious onset and, if untreated, can last up to several months. Upon resolution the sclera may look bluish due to a rearrangement of the collagen fibrils, with no loss of tissue or thinning. About 45% of patients with anterior diffuse scleritis will have an associated disease, RA been the most common.7–9
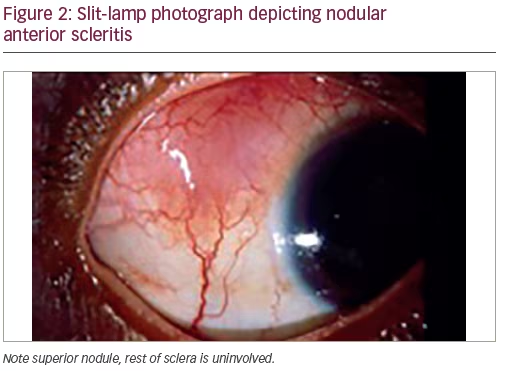
Nodular anterior scleritis (Figure 2)—this type of anterior scleritis is localized to a scleral nodule that is immobile, elevated, and firm. The nodule has a violaceous color with a congested vascular network. 40–50% of patients with nodular scleritis have an associated disease and RA is the most common. Patients with nodular scleritis may progress to anterior necrotizing scleritis and this needs to be carefully monitored.7,8
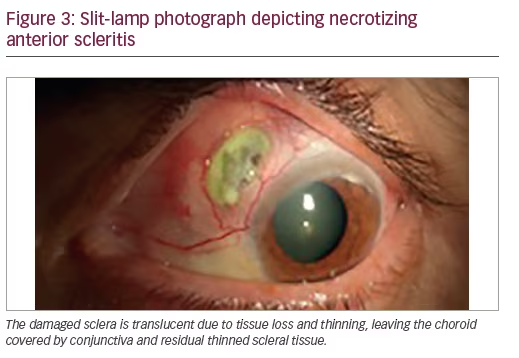
Necrotizing anterior scleritis (Figure 3)—necrotizing scleritis is the most severe form of scleritis and causes a significant amount of ocular morbidity. This form of scleritis is also a sign of the onset of a potential lethal systemic vasculitis.
This condition has an older age of onset and a higher proportion of patients (60–90%) have an underlying systemic disease, most commonly granulomatosis with polyangiitis and RA.9
The onset of necrotizing scleritis is gradual (3–4 days) and associated with severe pain. The affected avascular scleral tissue will look white and will be surrounded by intense swelling and redness of actively inflamed tissue. The inflammation starts in a demarcated area and will spread circumferentially and involve the whole anterior segment. The damaged sclera will become translucent due to tissue loss and thinning, leaving the choroid covered by conjunctiva or residual thinned scleral tissue. The protrusion of choroid can occur with trauma or increased intraocular pressure.7
It is important to keep in consideration that infections can also be a cause of necrotizing scleritis and need to be included in the differential diagnosis. Systemic immunosuppression is required for the treatment of anterior necrotizing scleritis associated with autoimmune diseases.
Scleromalacia perforans—This form of necrotizing scleritis without inflammation is almost always seen in patients with long-standing RA. It is characterized by the painless and slow disappearance of the overlying episcleral tissue, associated with attenuation of the conjunctival and episcleral vessels. The scleral tissue changes color from white to yellow and this becomes absorbed and disintegrated, leading to exposure of the underlying choroid. Although spontaneous perforation is rare, traumatic perforations can easily occur.7,8,10
Posterior scleritis
The onset of posterior scleritis has very few, and in some instances no physical signs, and the diagnosis can be challenging. Moreover, because posterior scleritis can present as a choroidal mass, serous retinal detachment, retinal striae or retinal and disc edema, it is confused with other diseases of the posterior segment. The most common presenting clinical feature is decreased
vision with pain and diplopia, flashes, and limitation of ocular movements can be present. Because of the close connection between the sclera and Tenon’s capsule, inflammation of the posterior sclera can extend to the orbit and cause proptosis, chemosis, lid swelling and retraction in upgaze. The diagnosis is made by B-scan ultrasonography, which will demonstrate thickening, edema of the posterior sclera, and a ‘T’ sign when edema of the Tenon’s space and the adjacent optic nerve occurs. The association of posterior scleritis with systemic disease is less compared to the anterior form, however, it is still significant and patients need to undergo a systemic evaluation.7–9,11
Diagnostic evaluation
The evaluation of a patient with scleritis requires a systemic evaluation. This should start with a thorough medical history with an extensive review of systems and a physical examination, in addition to a full ophthalmic examination. Infectious etiologies also should be considered and a history of trauma or surgical insult should be sought.10,11
Approximately 50–60% of patients with scleritis will have an underlying associated disease (see Table 1). Of these, 50% will be an autoimmune connective tissue or vasculitic disease and 50% of the patients presenting with necrotizing scleritis will have a mortality rate of 50–60% within five years of onset of disease, if not properly immunosuppressed. Therefore, early diagnosis and treatment is critical for a good ocular and systemic prognosis of patients with scleritis.
Laboratory tests for suspected systemic diseases in patients with scleritis must be target-oriented, based on the data generated from a comprehensive medical and ophthalmic examination. It should be emphasized that many systemic diseases do not have specific laboratory tests, and that diagnosis can only be made on the basis of the clinical and biological findings.
All patients with a new diagnosis of scleritis should undergo evaluation for the presence of a systemic vasculitis. Routine testing typically includes complete blood count, complete metabolic panel, urinalysis with microscopic analysis, perinuclear and cytoplasmic anti-neutrophil cytoplasmic antibody, and chest X-ray. Infections including syphilis and Lyme disease should be ruled out with a rapid plasma reagin test, fluorescent treponemal antibody, and a Lyme antibody.1,8–11
More directed evaluations may be ordered based on the history and physical examination. Most patients with RA and systemic lupus erythematosus carry their diagnosis prior to presenting with scleritis and, therefore, obtaining an antinuclear antibody or rheumatoid factor level may not be necessary unless dictated by the history and physical examination. Other potential directed tests may include a tuberculin skin test, sacroiliac joint X-rays (for spondyloarthropathy), sinus imaging (for Granulomatosis with polyangiitis), and viral hepatitis panel (hepatitis B for polyarteritis nodosa and hepatitis C for cryoglobulinemia). In any case of suspected infectious scleritis, cultures and/or scleral biopsy may be needed to secure the diagnosis.1,8–11
Pathology/pathogenesis
The pathology and pathogenesis of scleritis are multifactorial and complex. Inflammatory responses in the sclera can be granulomatous or nongranulomatous. This is in part determined by the cause of the scleritis, which can be infectious or autoimmune.
Although rare, infectious scleritis results from the direct invasion of the infectious agent, which triggers an inflammatory response and local tissue damage. Organisms that have been associated with scleral infections include herpes, syphilis, mycobacterium, acanthamoeba, bacteria (pseudomona), and fungi.1,11
In autoimmune disorders, a hypersensitivity reaction is generated against autoantigens, which leads to a cellular immunological attack against healthy tissue and vessels. The immunological mechanisms involved in scleritis are described by the Type III (immune-complex mediated) and Type IV (cell mediated) hypersensitivity reactions that leads to inflammatory microangiopathy and direct cellular damage of affected scleral tissue and vessels. Vessel occlusion and ischemia contribute to tissue damage and necrosis. The response to the inflammatory insult results in the activation of local mechanisms that lead to the degradation of proteoglycans and collagen, which eventually results in the thinning and loss of scleral tissue.11
Treatment
The treatment of scleritis requires the use of systemic immunomodulatory therapy (IMT). A step-ladder approach should be instituted for the treatment of scleritis and this can be adjusted depending on the severity of the presentation and specific diagnosed systemic disease. Diffuse, nodular or posterior scleritis can be initially treated with systemic non-steroidal anti-inflammatory drugs (NSAIDs) following the medical and pharmacological recommendations. Glucocorticoids should be used when failure of NSAIDs occurs or in cases when rapid control of destructive inflammation is needed. Based upon our clinical experience, we recommend initial therapy with prednisone 40–60 mg/day. The use of prednisone should be limited and chronic use needs to be avoided. Usually this regimen is continued for the first four weeks of therapy with ongoing assessment of clinical response.
If failure or chronic dependence to prednisone is developed, the use of immunosuppressive therapy drugs IMT may be added or substituted as third-line therapy. Moreover, IMT may be the initial choice in necrotizing scleritis.12
There are no randomized trials in scleritis on which to base the choice of the specific immunosuppressive medication. Also, new IMT and biologic response modifiers (BRM), mainly antitumor necrosis factor alfa (TNFα), rituximab (RTX) and adalimumab, may be effective in refractory scleritis.12–18
Systemic steroid-sparing agents that have been used with success in the treatment of necrotizing and chronic scleritis include methotrexate, azathioprine, cyclosporine, cotrimoxazole, mycophenolate mofetil, and cyclophosphamide.
Because of the proven effectiveness of both RTX and cyclophosphamide in patients with granulomatosis with polyangiitis, the first-line immunosuppressive medication in the treatment of scleritis is typically one of these agents.12,14,19–21
RTX is a chimeric mouse monoclonal antibody that directly targets the CD20 antigen expressed on the majority of B cells. There is increasing evidence that RTX can be employed to successfully treat ocular inflammatory disease.13 Given the frequency of severe scleritis as a manifestation of granulomatosis with polyangiitis and the strong evidence that RTX is effective in this setting, it is reasonable to extrapolate the efficacy of RTX to scleritis. The use of cyclophosphamide or RTX as a first line therapy should be considered in necrotizing scleiritis associated with systemic vasculitis, as this will also decrease the risk of death in these patients. For patients with disease refractory to RTX, we suggest cyclophosphamide (2 mg/kg per day, with dose adjustments for patients with decreased renal function). Progressive scleral melting will require scleral grafting surgery and systemic chemotherapy.13,16
Case reports and uncontrolled case series suggest that the TNFα inhibitor infliximab may be partially effective in the treatment of scleritis that is resistant to treatment with other agents. Doses in the range of 3–5 mg/kg administered every 4–8 weeks have been employed for this purpose.15–18
Surgical intervention is uncommon, but may be necessary for diagnosis, repair of scleral or corneal defects, or prevention of globe perforations
Infectious scleritis
Infectious scleritis can be viral, bacterial, fungal, and parasitic. It is uncommon, particularly in the absence of infectious keratitis; however, the overall visual outcome in infectious scleritis is generally worse than its autoimmune counterparts, perhaps because of the delay in diagnosis or because of the aggressive nature of associated microbes.22 Sometimes it is not easy to discriminate infections from inflammatory diseases, and clinical history and temporal profile become essential: infectious scleritis is usually acute and autoimmune scleritis may be a recurrent disease.
Many organisms have been reported as possible causes of scleritis. In a large series of 97 patients with scleritis over a 12-year period, 7.5% had an infectious disease and the most common infection was herpes zoster ophthalmicus.23
Infections of the sclera are often difficult to manage and eradicate because of the poor antimicrobial penetration into the avascular necrotic sclera, but improved success has been achieved with surgical intervention in addition to antimicrobial therapy, or a combination of parenteral antimicrobials.
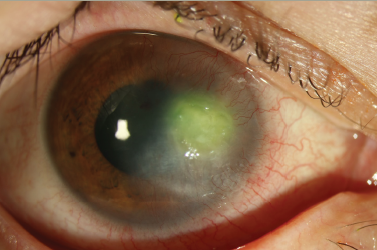
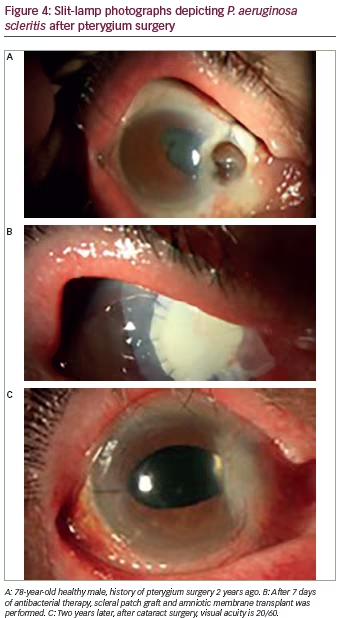
Infections occur in tissue compromised by disease or trauma, both iatrogenic and accidental. Surgery and history of ocular trauma are the most important risk factors. Scleral infection can commence a few weeks after anterior segment surgery or arise decades later. Pterygium excision seems to be the most common procedure predisposing to microbial scleritis, but cases after cataract extraction, trabeculectomy and pars plana vitrectomy have also been described.22 Although not a prerequisite for scleral infection, adjunctive use of irradiation or antimetabolites, specially mitomycin-C adds to the risk of scleral thinning and avascular necrosis that provide a nidus for microbial adherence. Necrotizing scleritis tends to be the most common presentation of infectious scleritis. Thus, infectious etiologies should be suspected in any case of progressive indolent necrosis with suppuration, refractory to anti-inflammatory regimens (see Figure 4).22
2013, our institution reported the epidemiology and outcomes of all patients with a positive microbial culture obtained by swab, spatula, or biopsy from sclera at Bascom Palmer Eye Institute from 1987–2010.5 Fifty-six eyes (55 patients) had confirmed infectious scleritis, which was defined as having a positive scleral culture. The median age at diagnosis was 70 years (range 5–92). Eighty-nine percent of eyes had an identifiable inciting factor associated with the development of scleritis. These included previous surgery (the most common being pterygium excision, typically with concomitant radiation or mitomycin C), followed by cataract extraction and trauma. Of 56 cases of infectious scleritis, 87% were due to bacterial species and 11% were due to fungi. Pseudomona aeruginosa was the most common causative organism isolated (n=20). Approximately 50% of eyes lost functional vision (worse than 20/200). Presenting visual acuity (VA) of worse than 20/200 and concomitant keratitis or endophthalmitis were associated with poorer VA outcomes.5
In 1991, Alfonso et al. published the results of 28 patients with culture proven infectious scleritis and keratoscleritis. Seven of eight patients who were treated with antibiotics alone and two of 11 patients who received surgical intervention and antibiotics eventually required evisceration or enucleation of the eye. Their results suggested that cryotherapy, lamellar, or penetrating corneoscleral grafts, in addition to intensive antibiotic therapy may improve the outcome of patients with infectious keratoscleritis.24
Effective treatment requires both aggressive medical and surgical methods. In addition to microbe-specific medical therapy, our group recommend debridement of scleral abscesses and necrotic tissue and cryotherapy if the sclera is not significantly damaged. We also recommend the use of topic and/or systemic steroids in bacterial infectious scleritis, especially in Pseudomona aeruginosa and Streptococcus cases. In fungal and mycobacterial infections, steroids are absolutely contraindicated.