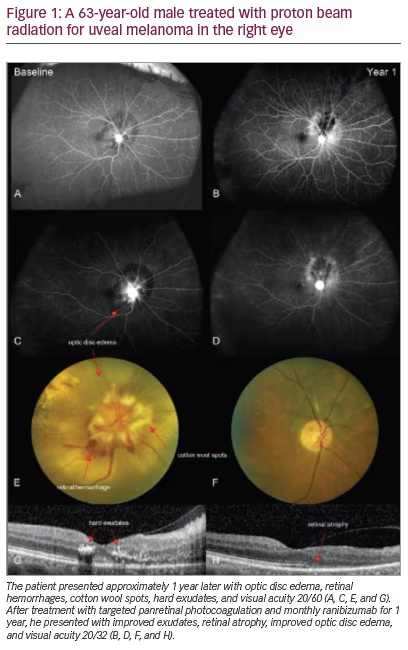
Uveal melanoma represents approximately 5–6 % of all melanoma diagnoses, the second most common form of melanoma.1 Uveal melanoma differs from cutaneous melanoma in many respects. Clinically, nearly every patient with uveal melanoma is diagnosed with disease limited to the primary site of disease in the uveal tract (see Figure 1). Uveal melanoma cells are thought to metastasize primarily by hematogenous spread due to a lack of lymphatic drainage in the uveal tract.2 When uveal melanoma metastasizes, the liver is involved in up to 95 % of patients, followed by the lungs (24 %), bone (16 %), and skin (11 %).3 Chemotherapy, biochemotherapy, and immunotherapy regimens attempted to date have failed to show significant efficacy in metastatic uveal melanoma.1,4
Uveal melanomas have a different cytogenetic profile compared with cutaneous melanomas, often containing alterations in chromosomes 1, 3, 6, and 8.5–9 Partial or total loss of chromosome 3 is believed to be an early genetic event and a strong predictor of metastatic disease. Chromosome 8q copy gains, which often occur in the same tumors that have chromosome 3 loss, portend a worse prognosis. Chromosome 6p gains are nearly mutually exclusive with chromosome 3 loss and are prognostic of a non-metastatic phenotype. More recently, microarray analysis performed by several investigators has revealed two distinct profiles,10,11 one associated with a low risk (‘class 1’) and another associated with a high risk (‘class 2’) of metastatic disease. In a retrospective study by Worley et al., univariate analysis showed class 2 gene expression, advanced patient age, and scleral invasion to be associated with metastasis; only class 2 gene expression was significantly associated with metastasis by multivariate analysis.12 No one gene in the class 2 profile was predictive of metastasis.
Although mutually exclusive BRAF or NRAS mutations are found in 60–70 and 20 % of cutaneous melanomas, respectively, multiple studies using standard techniques have failed to identify these mutations in uveal melanoma tissues.13–17 Using highly sensitive techniques, BRAF mutations have been identified in a subset of uveal melanomas, but appear to be present in small populations of cells within a tissue.18–20 Likewise, KIT mutations, which are found in approximately 15–20 % of acral lentiginous and mucosal melanomas,21 are not found in uveal melanoma tissues.22,23 Recent reports have demonstrated that uveal melanomas harbor activating GNAQ or GNA11 mutations in up to 85 % of cases.24,25 Thus it is clear that uveal melanoma differs molecularly and clinically from most other subtypes of melanoma. These differences present an opportunity and challenge for the future development of therapeutic interventions.
Uveal Melanocyte Development
Melanocytes are derived from pluripotent neural crest cells. Melanocyte precursors migrate out of neural crest and populate different anatomical locations, such as the epidermis, dermis, and uveal tract. Using mice with varying degrees of KIT receptor tyrosine kinase signaling activity due to KIT genetic aberrations or KIT receptor blockade, Aoki et al. showed that melanocytes in the dermis and uveal tract are less dependent on KIT signaling compared with those in the epidermis.26 Conversely, dermal and uveal tract melanocytes are highly dependent on endothelin-3 (ET3) and hepatocyte growth factor (HGF) signaling. These findings are consistent with those of Van Raamsdonk et al., who demonstrated that activating mutations in the human homologs of the G-protein αq (GNAQ) and G-protein α11 (GNA11) result in dermal melanocytosis (uveal tract melanocytes were not investigated in this study).27 Both GNAQ and GNA11 are coupled to the endothelin B receptor (EDNRB), for which ET3 is a ligand. These studies implicate the ET3/EDNRB–GNAQ/11 signaling pathway as important for the differentiation and growth of melanocytes in the dermal and uveal tract compartments.
Signaling Pathways in Uveal Melanoma
The identification of activating GNAQ and GNA11 mutations and elevated expression of insulin-like growth factor-1 receptor (IGF1R) and hepatocyte growth factor receptor (MET) receptor in uveal melanomas present the intriguing possibility that uveal melanoma cells may have signaling dependencies more akin to precursor uveal melanocytes. The MEK/mitogen-activated protein kinase (MEK/MAPK) and phosphatidylinositol-3 kinase (PI3K)/AKT pathways are major mediators of these upstream signals (see Figure 2). Importantly, clinically relevant small-molecule inhibitors have been developed for multiple molecules in the MEK/MAPK and PI3K/AKT pathways.
G-protein Alpha-q and G-protein Alpha-11 Mutations
Following the observation that activating mutations in GNAQ or GNA11 result in dermal melanocytosis in mice, van raamsdonk and Bastian investigated the GNAQ and GNA11 mutation status in subtypes of blue nevi, an intradermal melanocytic proliferative condition.24,28 They observed recurrent, mutually exclusive mutations in R183 and Q209 in both GNAQ and GNA11. GNAQ (Q209) mutations were present in 55 % of blue nevi, whereas GNA11 (Q209) mutations were observed in 7 %. GNAQ and GNA11 (R183) mutations were only present in 2 % of blue nevi. Recognizing that the dermally derived nevus of ota is a risk factor for uveal melanoma, this group also examined uveal melanoma tumors GNAQ or GNA11 (Q209) mutations were observed in 45 and 32 % of primary uveal melanomas, respectively. R183 mutations in either GNAQ or GNA11 were present in 6 % of primary uveal melanoma tumors. Stable expression of GNAQ or GNA11 (Q209L) in melanocyte cell lines transformed these cells and induced highly pigmented tumors when injected into mouse subcutaneous tissue. Exogenous expression of mutant GNAQ or GNA11 (Q209L) increased extracellular signal-related kinase (ERK)1/2 phosphorylation, suggesting an important role for the MEK/ERK pathway in GNAQ/11 mutant uveal melanoma.
Additional investigations into the frequency of GNAQ mutations in uveal melanoma have been performed. Onken and Harbour examined 67 primary uveal melanoma tissues and observed a 49 % GNAQ (Q209) mutation frequency.25 Bauer et al., showed a 53.3 % GNAQ (Q209) mutation frequency in 75 primary uveal melanomas.29 Finally, Dratviman-Storobinsky et al., identified 45.5 % of 27 primary and 36.5 % of 11 uveal metastases to harbor GNAQ (Q209) mutations.30 Three matched pairs of primary and metastatic tissues demonstrated GNAQ (Q209) mutations, suggesting this mutation to be an early event in uveal melanoma progression.
No statistically significant correlation has been observed between GNAQ or GNA11 mutation in the primary uveal melanoma tumor and clinical (overall survival, metastasis-free survival, sex, age), tissue (tumor thickness, diameter, pigmentation, or extracellular matrix patterns), cytogenetic (e.g., chromosome 3 status, 8q gain, 6p gain, 8p loss), or molecular (β-catenin, e-cadherin, cytokeratin-18) variables.25,30 GNA11 mutations were observed to be more prevalent in ciliochoroid tumors.
These data convincingly demonstrate that GNAQ and GNA11 are oncogenes in uveal melanoma, and targeting these genes (or protein products) or downstream signaling mediators may be a viable treatment strategy.
BAP1 Loss
It has long been postulated that the correlation between primary uveal melanoma tumors with monosomy 3 and the risk of metastatic disease may be accounted for through loss of heterozygosity of a locus on the remaining intact chromosome 3. The identification of gene aberrations that result in loss of BAP1, a previously described tumor suppressor located on chromosome 3p21.1, in a large number of primary uveal melanoma tumors with monosomy 3 provided strong evidence that BAP1 loss may be a very important mediator of metastatic disease.31 Of tumors that were monosomy 3, and for which BAP1 mutant status could be determined, BAP1 mutations were present in 81% of cases.
BAP1 is a nuclear ubiquitin carboxy-terminal hydrolase. Previous studies have shown BAP1 to have tumor suppressor activity.32,33 For example, BAP1 regulated cell proliferation by e-ubiquitinating HCF-1, a cell cycle regulator.32 A recent study shows BAP1 to be the previously uncharacterized drosophila gene calypso which is part of the polycomb group repressive de-ubiquitinase complex involved in removal of monoubiquitin from histone H2A, and ultimately stem cell pluripotency and organismal development.34 The exact mechanism by which loss of BAP1 mediates primary uveal melanoma metastasis is currently being investigated. A further understanding of these mechanisms may provide potential pathways that can be therapeutically targeted.
Hepatocyte Growth Factor and Hepatocyte Growth Factor Receptor
Given the propensity of uveal melanoma to metastasize to the liver, many investigators have hypothesized that the liver microenvironment may contain factors that are hospitable to uveal melanoma growth. HGF is highly produced in the liver and, when bound to the tyrosine kinase receptor MET, results in proliferation, survival, and migration in many cell types.35 As such, multiple groups have examined the expression level of MET in primary uveal melanoma tissues and cell lines. The results of these studies vary depending on the methods of analysis employed.
Economou et al. examined 132 primary uveal melanoma samples and showed 86 % of the tissues to have MET expression.36 The intensity of MET staining observed was approximately evenly divided into thirds: 30 % weak (<10 % staining), 27 % moderate (10–50 % staining) and 30 % strong (50–100 % staining). MET expression was associated with uveal melanoma-specific mortality by univariate, but not multivariate, analysis in this study. Using a different method of analysis, Mallikarjuna et al. reported 57 % of 60 primary uveal melanoma tissues to have MET expression. In this report, cells were divided into either heterogeneous (bright staining ≤10 %, dull staining >10 %), positive (bright staining >10 %), or negative.37 Staining was noted to be membranous. Positive staining was observed in 43 % and heterogeneous staining was observed in 23 % of tissues. Tissue with less than 10 % of cells showing ‘dull’ staining was counted as negative, which may account for the discrepancy between this and the Economou study. Importantly, Mallikarjuna et al. determined that primary tumors that metastasized showed a higher expression of MET than tumors that did not metastasize. There was a significantassociation between MET expression in primary uveal melanoma tissues and uveal melanoma-specific mortality. Abdel-Rahman et al., also using a different method of analysis, found 96 % of 46 primary uveal melanomas to have MET expression.38 In this analysis, the percentage of stained cells multiplied by the intensity of staining(0–2) was used to determine whether tissues were negative (<10), weak/moderate (10–100), or strong (>100). Strong staining was observed in 57 % and weak/moderate staining was observed in 39 % of tissues. Localization of staining was stated as being mostly cytoplasmic. Finally, Topcu-Yilmaz et al. recently reported only 12.5 % of 40 primary uveal melanoma tissues to have MET expression. This analysis only considered cells with membrane staining to be positive.39
Differences observed in these studies are probably accounted for by divergent methods and analyses. However, these studies provide convincing evidence that MET receptor expression and function merit further study in uveal melanoma pathogenesis. Abdel-Rahman et al. examined the effect of the small molecule MET inhibitor SU11274 on cell proliferation and migration of uveal melanoma cell lines. Elevated phosphorylation of MET corresponded with total MET expression in the cell lines examined.38 Notably, the three cell lines that lacked significant MET and p-MET expression are known to harbor BRAF mutations, which have only been observed in a few cases of uveal melanoma using the ultrasensitive techniques19 (and our unpublished data). Treatment of serum-starved uveal melanoma cell lines with SU11274 diminished the phosphorylation of MET. When compared with normal retinal pigmented epithelium or fibroblasts, uveal melanoma cells exhibited a lower IC50 value when treated with SU11274, but differences among the uveal melanoma cell lines were not statistically significant. Treatment with SU11274 also inhibited uveal melanoma cell migration in vitro.
Insulin-like Growth Factor and Insulin-like Growth Factor Receptor
Like HGF, the insulin-like growth factor-1 (IGF-1) is highly expressed in the liver and has been shown to mediate proliferation, survival, and migration in many cell types.40 Economou et al. examined 132 primary uveal melanoma samples and showed 73 % of the tissues to have IGF1R expression.36 Using the same analysis method used for MET analysis, they observed the intensity of IGF1R staining to be as follows: 42 % weak (<10 % staining), 22 % moderate (10–50 % staining), and 10 % strong (50–100 % staining). The relative overlap between IGF1R and MET expression was not determined. Multivariate analysis showed elevated IGF1R expression to be associated with melanoma-specific mortality. All-Ericsson et al. examined 36 primary uveal melanoma tissues using similar criteria and observed a 94 % frequency of IGF1R expression.41 Topcu-Yilmaz et al., limiting IGF1R positivity to membrane-only staining, observed a lower frequency of 20 % of the 40 uveal melanoma tissues examined.39 As in the case of MET, these studies provide convincing evidence that IGF1R expression and function merit further study in uveal melanoma pathogenesis.
Using an IGF1R inhibitor, cyclolignan picropodophyllin (PPP), Girnita et al performed in vitro and xenograft experiments using uveal melanoma cell lines. Treatment with PPP diminished phospho-IGF1R levels, decreased cell viability, and inhibited cell migration.42 In addition, tumor volume was dramatically reduced after PPP treatment in a uveal melanoma xenograft. Notably, the majority of in vitro and all the xenograft experiments in this study were performed with uveal melanoma cell lines known to harbor a BRAF mutation.
MEK/Mitogen-activated Protein Kinase Pathway
Activation of the MEK/MAPK pathway provides an essential proliferation signal in many tumor types. This pathway is clearly activated in a majority of uveal melanoma tissues and cell lines. Zuidervaart et al. showed that half of 19 tissues examined had >50 % cells staining for phosphorylated MAPK with no tissues being negative for phosphorylated MAPK.43 Weber et al. observed 86 % of 42 uveal melanoma tissues to have nearly homogeneous staining for phosphorylated MAPK.13 Positivity did not correlate with cell type (epithelioid, spindle, or mixed) or location (ciliary body or choroid). Multiple groups have shown that chemical blockade of MEK in uveal melanoma cells results in the loss of MAPK phosphorylation, decreased cell proliferation, and the induction of apoptosis.24,44
Phosphatidylinositol-3 Kinase/AKT Pathway
Activation of the PI3K/AKT pathway provides an important survival signal in many tumor types. Saraiva et al. examined 18 primary enucleated choroid melanoma specimens and 16 specimens that had undergone both radiotherapy and enucleation and observed a 55.5 and 56.2 % phosphorylated AKT expression, respectively.45 Activation of AKT in the enucleated-only group was associated with a higher risk of metastatic disease. Populo et al. identified 56.7 % expression of phospho-AKT (Ser473) and 55.2 % of phospho-AKT (Thr308) in the 30 uveal melanoma samples they examined.46 A statistically elevated level of phospho-AKT (Thr 308) was observed in uveal melanomas that had metastasized; however, the number of samples was limited.
Phosphatase and tensin homolog (PTEN) acts as a tumor suppressor primarily by converting inositol triphosphates into inositol bisphosphates, thus modifying the effect of active PI3K. Mutations, deletions, or epigenetic inactivation of PTEN result in an enhanced PI3K effect and increased cell survival. Abdel-Rahman et al. demonstrated that 76.3 % of 38 primary uveal melanomas exhibited a loss of heterozygosity and 11.4 % of 35 tumors had a mutation in the coding region of PTEN.47 Loss of cytoplasmic PTEN expression was associated with shortened disease-free survival.
Babchia et al. treated uveal melanoma cells with the reversible pan-PI3K chemical inhibitor LY294002. Inhibition of PI3K resulted in a marked reduction in cell proliferation and induction of apoptotic cell death in a majority of cells.44
Conclusions
Recent identification of cytogenetic abnormalities and their associated clinical relevance has marked a major accomplishment in the biological understanding of uveal melanoma. With the identification of GNAQ, GNA11 and BAP1 mutations and important signaling pathways, we now enter a new era in uveal melanoma research. Understanding and integrating genetic/epigenetic profiles with the signaling pathways that mediate uveal tumor growth, survival, and metastasis is now a key aim of many research investigations. The challenge will be to translate this knowledge into effective therapeutics in the adjuvant and advanced uveal melanoma settings.
Important questions remain to be answered. Despite more refined chromosomal analyses showing chromosomal segment losses or gains, the concrete identification of particular genes or epigenetic alterations that mediate disease has not been elucidated. Systems-based approaches that investigate the interplay of genetic and epigenetic alterations will be necessary to determine their pathological relevance. Similarly, the functional significance and potential redundancy in cellular signaling cascades will need to be parsed in order to determine optimal therapeutic targets. Although chemical inhibitor studies performed to date have provided clues to important signaling processes, experiments employing clinically relevant small-molecule inhibitors in cell lines and animal models that reflect the genotypic profile of uveal melanoma tissues are necessary. Designing clinical trials specific to uveal melanoma patients is challenging due to limitations in the number of patients and a historically low level of commitment to drug development specific to uveal melanoma, due to the relative rarity of this melanoma type. However, the recent identification of signaling pathways that may be relevant in the pathogenesis of metastatic uveal melanoma, coupled with the recent ability to identify patients at high risk of developing metastasis, creates an opportunity to design trials that use a personalized targeted therapy approach.