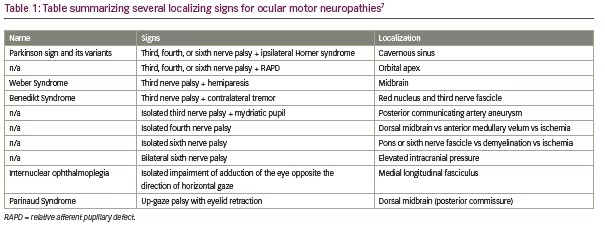
The Parkinson sign is classically described as a sixth nerve palsy and an ipsilateral Horner syndrome that localizes to the cavernous sinus. 1,2 The localizing value of any afferent and efferent (e.g., Horner syndrome) pupillary finding with an ipsilateral ocular motor cranial neuropathy merits revisiting the Parkinson sign.3,4 For example, a unilateral pupil-sparing cranial nerve III palsy with an ipsilateral smaller pupil (rather than a larger pupil), a normal pupillary light reaction, an anisocoria worse in the dark (ocular sympathetic pupil dilation problem), and absence of aberrant regeneration suggests a concurrent ipsilateral Horner syndrome. Like the Parkinson sign, this combination of a third nerve palsy and a Horner syndrome localizes to the ipsilateral cavernous sinus. The classic Parkinson sign, as a localizing finding to the cavernous sinus, thus might be extended to include any ocular motor cranial neuropathy (e.g., III, IV, or VI) in conjunction with an efferent oculosympathetic pupillary defect (e.g., Horner syndrome). In contrast, a patient with a relative afferent pupillary defect and a concomitant ocular motor cranial neuropathy localizes just anterior to the cavernous sinus in the orbital apex. We believe that the Parkinson sign should be revisited as a valuable reminder of the localizing value of any combination of afferent and efferent pupillary problems in ocular motor cranial neuropathy.
Case 1
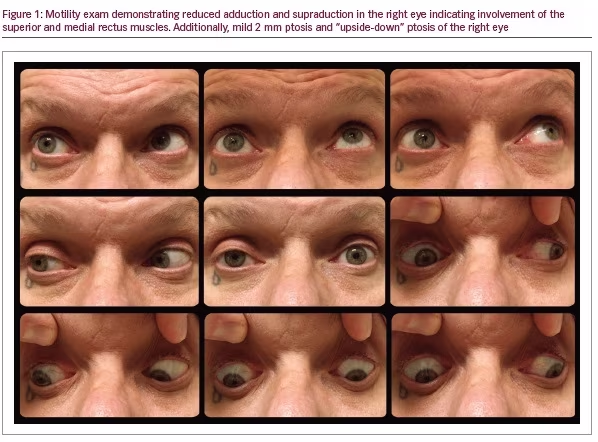
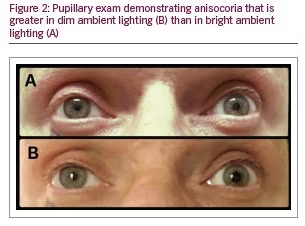
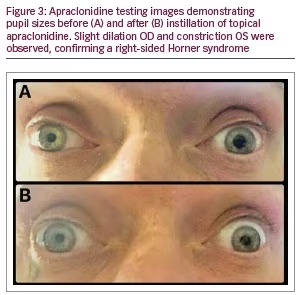
A 43-year-old male presented with 1 year of progressively worsening binocular horizontal diplopia and right retro-orbital pressure. Past medical, surgical, family, and social histories were unremarkable and he was taking no medications. Visual acuity measured 20/25 in each eye (OU). Slit lamp and dilated fundus exams were normal. The patient had 2 mm of upper eyelid ptosis and “upside-down” ptosis (lower lid elevation, often secondary to sympathetic denervation of the lower lid retractor) of the right eyelid and impairment of adduction and supraduction in the right eye consistent with a partial third nerve palsy (Figure 1). Pupil exam showed a normal light reaction OU and an anisocoria greater in the dark. Pupils measured 3 mm in the right eye (OD) and 5 mm in the left eye (OS) in the dark, and 2 mm OU in the light. A miotic right pupil and a dilation lag OD were seen (Figure 2). The right pupil dilated after topical administration of apraclonidine eye drops consistent with a right Horner syndrome (Figure 3).
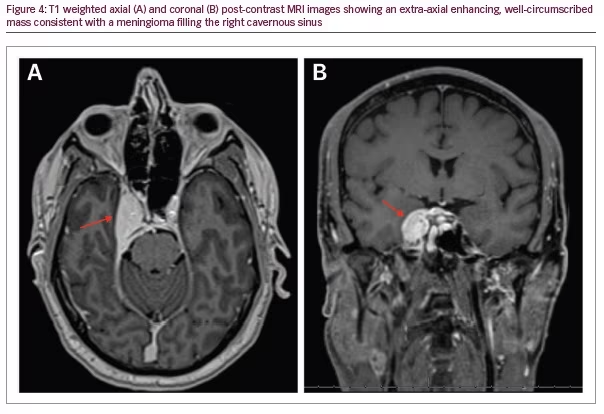
Magnetic resonance imaging (MRI) of the brain and orbits showed a right para-sellar, extra-axial mass filling the right cavernous sinus and Meckel’s cave (Figure 4). The mass encased the cavernous portion of the internal carotid artery and abutted the optic chiasm inferiorly without displacement. It extended into the sella, leftwardly displacing the pituitary gland and infundibulum, and there was suspected trans-osseous invasion of the right sphenoid sinus. The patient was not a candidate for surgical biopsy; however, a presumed diagnosis of meningioma was made due to its extra-axial location, involvement of Meckel’s cave, encasement of the right internal carotid artery, iso-intense character with respect to grey matter on T1 and T2-weighted imaging, and post-contrast enhancement. The differential diagnosis still includes schwannoma, plexiform neurofibroma, or cavernous hemangioma.5 The patient was referred to radiation oncology for possible radiation or chemotherapy with the goal of tumor size reduction.
Case 2
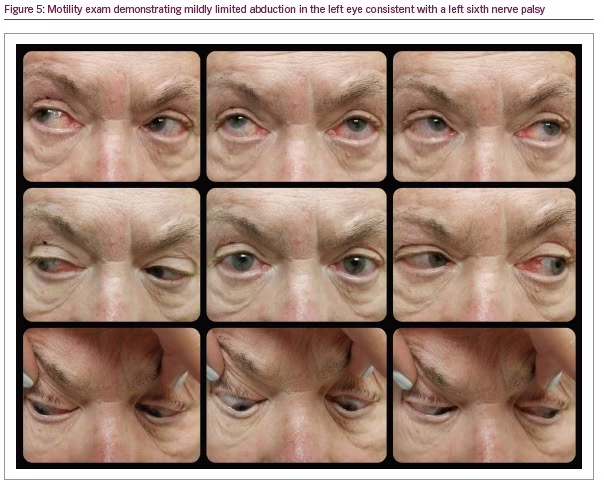
A 70-year-old male presented with transient visual loss in the OS and binocular horizontal diplopia. Past medical history was significant for Lynch syndrome, as well as renal cell carcinoma and sclerotic inflammatory obliterative thrombophlebitis with secondary pancreatitis, with a focus of low grade pancreatic intraepithelial neoplasia. His medications included insulin, antihypertensives, atorvastatin, and pantoprazole. Review of systems was negative for symptoms of giant cell arteritis. He had a strong family history of cancer and his social history was non-contributory. Visual acuity measured 20/25 OD and 20/20 OS. Both pupils measured 2 mm in the light and 4 mm in the dark, with a left relative afferent pupillary defect (RAPD). Slit lamp and dilated fundus exams were normal OU. Extraocular eye movements revealed limitation of abduction of the left eye consistent with left sixth cranial nerve palsy (Figure 5). The patient had a normal visual field.
A lesion involving the left orbital apex was suspected, but computed tomography (CT) and MRI revealed no mass. The patient subsequently suffered a transient ischemic attack; stroke workup including CT angiogram was performed which showed scattered calcifications in the bilateral cavernous to supraclinoid internal carotid artery but no lesion to account for the sixth cranial nerve palsy. Laboratory studies revealed erythrocyte sedimentation rate (ESR) of 60 mm/hour (normal ESR level for his age is <35 mm/hour) and a C-reactive protein (CRP) >30 mg/L (normal CRP level is <3 mg/L), and a temporal artery biopsy was positive for giant cell arteritis.
Discussion
Parkinson sign (Horner syndrome and an ipsilateral sixth cranial nerve palsy) is a well-known localizing sign to the cavernous sinus.1,2 However, the localizing value to the cavernous sinus applies to any ocular motor cranial neuropathy with a concomitant Horner syndrome. In addition, an afferent pupillary defect, in conjunction with an ipsilateral ocular motor cranial neuropathy, localizes to the ipsilateral orbital apex.3,4
Horner syndrome is caused by disruption of the sympathetic pathway, which starts in the hypothalamus, descends posterolaterally through the brainstem to the ciliospinal center of Budge (C8-T2), travels upward to synapse at the superior cervical ganglion, passes through the cavernous sinus with the internal carotid artery, and exits with the V1 branch of the trigeminal nerve to innervate the pupillary dilator, Müeller’s muscle, and the lower lid retractor.6 The presence of an ipsilateral Horner syndrome in Case 1 further localized the lesion to an area where the paths of both cranial nerve III and the sympathetic chain are close enough to be affected by the same lesion. This only occurs in the midbrain and the cavernous sinus. The absence of signs or symptoms a brainstem lesion and concomitant nerve palsies confirmed localization to the cavernous sinus. A midbrain cause can also be ruled out by confirming a third-order Horner syndrome via hydroxyamphetamine testing, which was not done here.
There are also specific scenarios for checking for both an efferent or afferent pupillary defect in patients with ocular motor cranial nerve palsy which may be vision or life-saving. For example, in a pupil-involved third cranial nerve palsy (with a fixed and dilated pupil) the RAPD can still be assessed using the fellow eye. Swinging the light from the affected (fixed and nonreactive pupil) ipsilateral eye to the uninvolved contralateral eye will cause pupil constriction to light in the normal eye. When the light swings back from the uninvolved normal pupil to the contralateral fixed and dilated pupil (the eye with the efferent pupillary defect) then observing the normal pupil for dilation can define a RAPD by “reverse testing”. Although it is referred to as a “reverse” RAPD test, the finding of dilation of both eyes upon swinging to the afferent affected eye occurs every time there is a RAPD regardless of the presence of the efferent pupillary defect. Thus, the RAPD test requires two eyes but only one working pupil. For a pupil involved third nerve palsy this would change the imaging strategy from evaluation for an aneurysm (e.g., posterior communicating artery aneurysm) to an evaluation of orbital apex syndrome (e.g., fungal disease in a diabetic patient).
In case 2, presented here, the patient had a RAPD (presumably due to optic neuropathy) and sixth nerve palsy. As this is evidence of both an efferent and afferent defect, this would normally localize to the orbital apex. However, despite history of Lynch Syndrome, our patient’s imaging was negative, demonstrating the potential false-localization of an ocular motor nerve palsy with a concomitant RAPD. In elderly patients this combination of afferent (RAPD) and efferent (ocular motor palsy) disease should prompt consideration for giant cell arteritis. The mechanism of diplopia in giant cell arteritis remains controversial but our second case suggests that it might be myogenic rather than neurogenic (orbital ischemic involvement from giant cell arteritis).
These cases provide a valuable reminder that a careful afferent and efferent pupil exam can be cross-referenced with an ocular motility exam, often allowing for localization at the bedside. Several combinations of ocular motor and pupillary or other exam findings have been described previously and are well-known to provide localizing value, including Parkinson sign, Weber Syndrome, Benedikt Syndrome, and Internuclear Ophthalmoplegia, among others (Table 1).7
In summary, we believe that the Parkinson sign should be revisited as a useful reminder of the role of a careful afferent and efferent pupil exam in cases presenting with diplopia and ophthalmoplegia.