Neovascular age-related macular degeneration (nAMD), diabetic macular oedema (DMO) and retinal vein occlusion (RVO) associated with macular oedema are some of the leading causes of visual impairment and loss worldwide.1–3 The pathogenesis of these diseases is largely driven by the family of pro-angiogenic vascular endothelial growth factor (VEGF) molecules, which are upregulated in response to vascular leakage and retinal oedema caused by retinal hypoxia.4 The standard of care for these conditions is intravitreal anti-VEGF injections, which not only limit disease progression, but also improve visual outcomes.5–8 Four anti-VEGF agents are approved by the US Food and Drug Administration (FDA) for intraocular use, including pegaptanib sodium (Macugen®, Gilead Sciences, San Dimas, CA, USA), ranibizumab (Lucentis®, Genentech, San Francisco, CA, USA), aflibercept (Eylea®, Regeneron Pharmaceuticals, Tarrytown, NY, USA), and most recently in 2019, brolucizumab (Beovu®, Novartis Pharmaceuticals, East Hanover, NJ, USA).9–12 Bevacizumab (Avastin®, Genentech, San Francisco, CA, USA) is also commonly used as an off-label treatment for these retinal diseases.13
While these biologic agents have revolutionized the treatment landscape for nAMD, DMO and RVO, there are several notable shortcomings with anti-VEGF therapy. With the chronic nature of these conditions, patients often require frequent injections and follow-up visits, which place a significant burden on patients, providers and the healthcare system. In addition, real-world studies have found that patients in routine practice are often undertreated and attain poorer long-term visual outcomes than those in clinical trials.14,15 Lastly, there always poses the risk for complications with each injection, such as intraocular inflammation, endophthalmitis or other non-ocular adverse events.16 Thus, alternative or adjunctive treatments to anti-VEGF monotherapy are needed to improve durability and efficacy.
One novel treatment target that has garnered recent interest is the angiopoietin-Tie-2 (Ang/Tie-2) pathway, which plays a key role in inflammation and vascular permeability.17 Faricimab (Vabysmo®, Genentech, San Francisco, CA, USA) is the only FDA-approved bispecific immunoglobulin designed for intraocular use to target both the VEGF-A and Ang/Tie-2 pathways in nAMD and DMO.18 With on-going phase III trials assessing intravitreal faricimab injections in RVO, it is proving to be a promising pharmacologic in the management of retinal pathologies.19,20 This review will overview faricimab’s design, the clinical trial evidence supporting its use in retinal vascular conditions and the on-going considerations for its use in clinical practice.
Overview of the angiopoietin-Tie-2 pathway and faricimab’s molecular design
The pathogenesis of vascular retinal diseases such as nAMD, DMO and RVO involves increased production of VEGF, a family of molecules that includes the four isoforms VEGF-A, -B, -C and -D.4 VEGF-A in particular has been a key therapeutic target due to its role in promoting angiogenesis and vascular permeability.21 For instance, the mechanism of action for widely used intravitreal injections such as ranibizumab, aflibercept and bevacizumab incorporates high-affinity inhibitory binding to VEGF-A to suppress abnormal blood-vessel growth.22–24
While VEGF-A plays a large role in new vessel growth, it interacts with a variety of regulatory pathways that contribute to vasculogenesis. One such pathway is the Ang/Tie-2 axis, which primarily regulates vascular health, permeability and inflammation.25 Tie-2 is a tyrosine kinase receptor found on vascular endothelial cells, and its two major physiological ligands are angiopoietin-1 (Ang-1) and angiopoietin-2 (Ang-2), which have opposing effects.26 Ang-1, normally expressed in pericytes, is an agonist that causes the phosphorylation of Tie-2 and activates downstream signalling that promotes vessel stabilization, anti-inflammation, pericyte health and vascular homeostasis.17 One such downstream pathway that reduces vascular permeability is by migration of the phosphorylated Tie-2 receptor to the cell junction, where it helps strengthen and stabilize the adherens protein vascular endothelial cadherin (VE-cadherin) that serves as a protective barrier against inflammatory stress and vascular permeability.27
In contrast, the role of Ang-2 is two sided; it is a partial agonist in normal homeostasis, but an antagonist in pathological conditions of the vascular endothelium such as in nAMD and diabetes.28 While the context-dependent nature of Ang-2 is still under investigation, serum Ang-2 levels are highly elevated in hypoxia, oxidative stress and hyperglycaemia.29 Pro-inflammatory environments, such as in nAMD, DMO and RVO, trigger the release of pre-formed Ang-2 from Weibel–Palade bodies within endothelial cells into serum.26
When serving as a competitive antagonist in pathologic states, Ang-2 blocks the phosphorylation of Tie-2, leading to vascular destabilization of the endothelium, promoting fluid leakage, and the expression of pro-inflammatory cytokines.30 While the exact mechanism of Ang-2’s effects is still under investigation, rat models with elevated Ang-2 expression have defective intra-endothelial adherens junctions, disrupted glycocalyx and upregulation of caveolin-1, which is associated with vascular permeability.31 In addition, in certain conditions, such as hyperglycaemia in diabetic retinopathy, Ang-2 enhances pericyte apoptosis, leading to breakdown of the blood–retinal barrier.32 Lastly, Ang-2 has a synergistic effect with VEGF in angiogenesis, since endothelial vascular permeability triples compared with VEGF alone.33,34 Thus, the interplay between VEGF and the Ang/Tie-2 pathway demonstrates how the pathogenesis of retinal diseases is often multifactorial, and requires a multifaceted treatment approach for enhanced efficacy.
Faricimab development and preclinical trial data
Faricimab is the first bispecific antibody administered intravitreally that binds to and inhibits both VEGF-A and Ang-2.35 The molecule was created with CrossMAb technology to allow heterodimerization of two antigen-binding fragments (Fab) onto one molecule. Faricimab is composed of an anti-VEGF-A Fab, an anti-Ang-2 Fab and a modified fragment crystallizable (Fc) region. The Fc region was engineered to eliminate its ability to bind with neonatal Fc and Fcγ receptors, decreasing systemic half-life and reducing inflammatory potential.35
The premise behind faricimab’s mechanism of action stemmed from a study of choroidal neovascularization (CNV)-induced mouse and non-human primate models.35 Neutralization of both VEGF-A and Ang-2 was more effective in reducing inflammation and vascular permeability than just targeting one factor.35 Thus, faricimab’s dual effects were hypothesized to be a novel option for patients refractory to anti-VEGF monotherapy, as well as to decrease the need for frequent treatment visits.26,36
The first phase I trial evaluating the tolerability and safety of faricimab enrolled 24 patients with nAMD and studied the effect of a single dose (SD) and multiple doses (MD).36 The SD component enrolled four cohorts with three patients each to receive faricimab 0.5 mg, 1.5 mg, 3 mg and 6 mg in a stepwise dose-escalation manner. Meanwhile, the MD component enrolled two cohorts with six patients each to receive monthly treatments of either faricimab 3 mg or 6 mg, for a duration of 3 months. Faricimab led to improvements in both visual acuity and optical coherence tomography features, without any dose-limiting toxicities. There were minimal ocular adverse events reported, all deemed unrelated to the study drug.36 With these promising results, further phase II and phase III trials were developed to investigate faricimab’s efficacy and safety not only in nAMD, but also in DMO and RVO.
Faricimab in neovascular age-related macular degeneration
STAIRWAY
STAIRWAY was a phase II, multicentre, comparator-controlled randomized study evaluating the efficacy of extended faricimab dosing compared with monthly ranibizumab in treatment-naive patients with nAMD.37 Enrolled patients were randomly assigned in a 1:2:2 ratio to receive ranibizumab 0.5 mg every 4 weeks (Q4W), faricimab 6 mg every 12 weeks (Q12W) or faricimab 6 mg every 16 weeks (Q16W). All patients in the faricimab arms were initiated on injections each month until Week 12. If eyes in the faricimab Q16W arm had signs of active disease at Week 24 by
pre-determined criteria, they were switched to Q12W dosing. The primary endpoint was mean change in best corrected visual acuity (BCVA) at Week 40, with secondary endpoints of anatomical improvements and safety.37
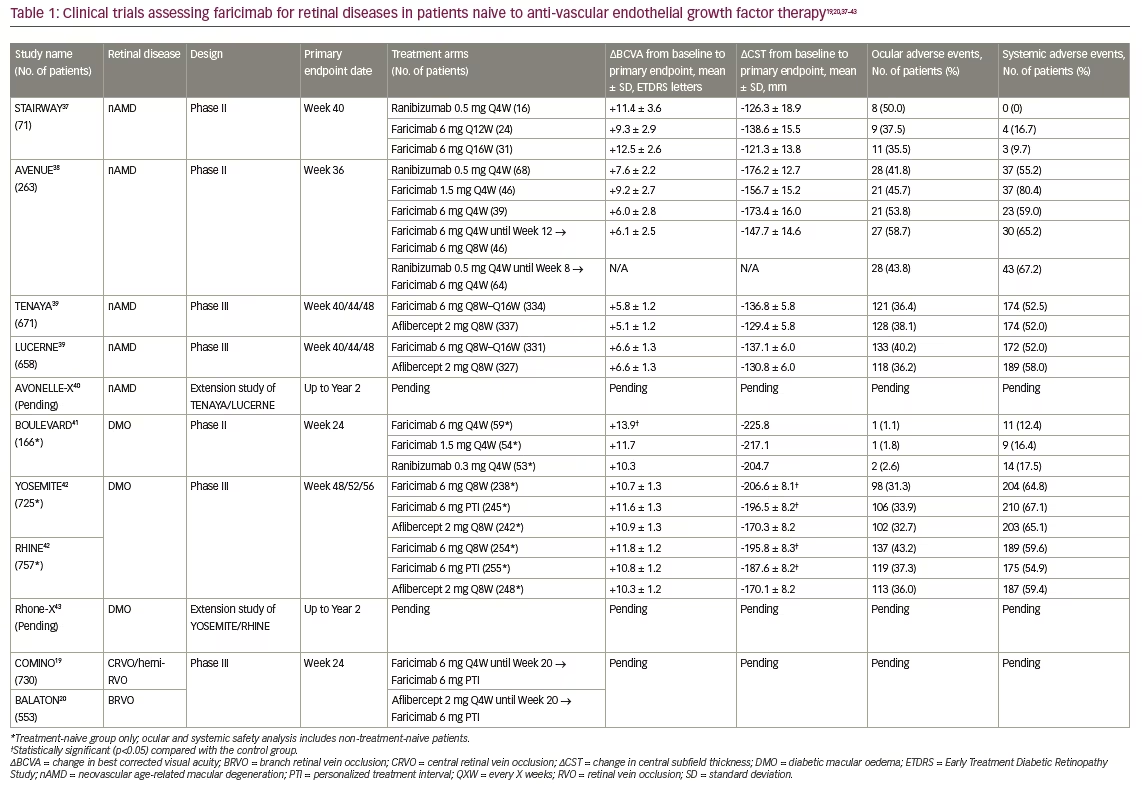
At Week 40, there were no clinically significant differences identified between the faricimab and ranibizumab treatment arms (Table 1).19,20,37–43 Secondary endpoints of mean changes from baseline in central subfield thickness (CST) and total lesion area were also comparable to ranibizumab treatment, demonstrating the ability of faricimab to address both visual and anatomical outcomes.37
AVENUE
AVENUE was another phase II, MD-regimen, comparator-controlled, randomized multicentre study assessing the efficacy and safety of faricimab compared with ranibizumab in treatment-naive patients with nAMD.38 Patients were randomized to five treatment arms in a 3:2:2:2:3 ratio. The first arm served as a control, with patients receiving ranibizumab 0.5 mg Q4W. The treatment arms for faricimab were 1.5 mg Q4W, 6 mg Q4W, and 6 mg Q4W until Week 12 followed by 6 mg every 8 weeks (Q8W). The last treatment arm was given ranibizumab 0.5 mg Q4W until Week 8, then faricimab 6 mg Q4W. The primary endpoint was mean change in BCVA at Week 36, with secondary endpoints analysing anatomic and safety parameters.38
At Week 36, all faricimab treatment arms had improved vision and anatomic features, such as CST reduction, similar to the monthly ranibizumab arm (Table 1). In addition, ocular and systemic safety findings for faricimab were comparable to monthly ranibizumab, with no serious adverse events considered related to study treatment. Although faricimab did not meet its primary endpoint of superiority over ranibizumab in the AVENUE study, its overall non-inferior visual and anatomic improvements warranted further trials.38
TENAYA/LUCERNE
With the promising phase II trial results, global phase III trials TENAYA and LUCERNE were conducted.39 TENAYA and LUCERNE were identically designed, multicentre, randomized, active comparator-controlled double-masked trials that assessed the sustained efficacy of faricimab in nAMD using individualized treatment intervals of up to Q16W. Patients recruited from 271 sites worldwide were randomly assigned in a 1:1 ratio to receive faricimab 6 mg or aflibercept 2 mg. Patients in the faricimab group were initially dosed with four Q4W injections up to Week 12. Following this loading phase, patients were assigned to Q8W, Q12W or Q16W dosing intervals based on assessments for active disease at pre-defined time points up to Week 60. Patients in the aflibercept group were initially dosed with three Q4W injections up to Week 8, followed by Q8W dosing up to Week 60. The primary endpoint was mean change in BCVA from baseline to Year 1, averaged over Weeks 40, 44 and 48, with a prespecified non-inferiority margin of four letters. Safety profile analyses included those who received at least one dose of study treatment.39
Both trials met their primary endpoints, with faricimab showing non-inferiority to aflibercept in both TENAYA and LUCERNE (Table 1). Furthermore, the lower bounds of the BCVA change differences between the treatment arms were well within the non-inferiority margin of four letters (TENAYA=0.7 letters; LUCERNE=0.0 letters).39 Faricimab treatment was durable, as 78% of patients in both studies achieved ≥Q12W dosing and 45% of patients reached the maximum dosing interval of Q16W by Year 1. Those treated with faricimab also demonstrated meaningful reductions in CST, comparable to the aflibercept group. The incidence of ocular adverse events was comparable across treatment groups and was consistent with expected events in patients receiving intravitreal nAMD treatment. Lastly, no cases of retinal vasculitis or retinal occlusive events were reported in either study.39
Year 2 data for both TENAYA and LUCERNE are currently being collected, with patients in the faricimab group receiving personalized interval dosing ranging from Q8W to Q16W up to Week 108, with a final visit at Week 112.39
AVONELLE-X
An extension study of TENAYA and LUCERNE, named AVONELLE-X, was launched to assess the long-term safety of faricimab in nAMD.40 AVONELLE-X is a multicentre, 2-year extension study designed to examine the long-term safety and tolerability of faricimab 6 mg in patients who had completed the TENAYA or LUCERNE clinical trials. This study is currently recruiting and is expected to conclude in August 2024.40
Faricimab in diabetic macular oedema
BOULEVARD
BOULEVARD was a phase II, randomized, active comparator-controlled, double-masked multicentre study that assessed the efficacy and safety of faricimab compared with ranibizumab in the treatment of DMO.41 Treatment-naive patients were randomized 1:1:1 to receive faricimab 6 mg Q4W, faricimab 1.5 mg Q4W or ranibizumab 0.3 mg Q4W, whereas patients previously treated with anti-VEGF therapy were randomized 1:1 to receive faricimab 6 mg Q4W or ranibizumab 0.3 mg Q4W. All patients were dosed monthly up to Week 20, followed by an off-treatment observation period up to Week 36 where investigators assessed treatment durability. The primary endpoint was mean change in BCVA from baseline to Week 24 in treatment-naive patients, with secondary endpoints of anatomic and safety outcomes.
The study met its primary endpoint, as patients receiving faricimab 6 mg had statistically significantly higher mean BCVA gains compared with those receiving ranibizumab (p=0.03; Table 1). In both treatment-naive and previously treated patient populations, faricimab treatment dose-dependently reduced CST and diabetic retinopathy severity scale scores, with longer time to re-treatment during the observation period compared with ranibizumab.41 There were no unexpected safety issues for faricimab during this study, and there were no reported adverse events of intraocular inflammation, endophthalmitis or retinal detachment. Findings from BOULEVARD supported that, compared with anti-VEGF monotherapy, bispecific targeting of Ang-2 and VEGF with faricimab extended durability in DMO.41
YOSEMITE/RHINE
Following the favourable results from BOULEVARD, two worldwide phase III studies were launched to assess faricimab in DMO.42 YOSEMITE and RHINE were two identical, randomized, double-masked non-inferiority trials that recruited both treatment-naive and previously treated patients. Patients were randomized 1:1:1 to receive faricimab 6 mg Q8W, faricimab 6 mg per personalized treatment interval (PTI) with adjustable dosing up to Q16W, or aflibercept 2 mg Q8W. Based on prespecified CST and BCVA criteria from Week 12 and onwards, patients in the PTI arm could extend their treatment intervals every 4 weeks (up to Q16W), or have these intervals reduced by 4 weeks or 8 weeks (as low as Q4W). All patients attended study visits Q4W and received sham injections at non-dosing visits to maintain masking throughout. The primary endpoint was mean change in BCVA from baseline to Year 1, averaged over Weeks 48, 52 and 56. Secondary endpoints included both anatomic and safety parameters. Safety analysis included all patients who received at least one dose of treatment.
Both YOSEMITE and RHINE met their primary endpoint, with the faricimab treatment arms demonstrating non-inferiority compared with the aflibercept arm (Table 1). In addition, more than 70% of patients in the PTI arm achieved a dosing interval of Q12W or greater at 1 year, highlighting the durability of faricimab. Anatomically, the faricimab arms showed significantly larger reductions in mean CST, with more patients achieving absence of DMO by Week 52 compared with the aflibercept arm. Lastly, faricimab was well tolerated, with ocular and systemic adverse events relatively low and well balanced across all treatment groups.42
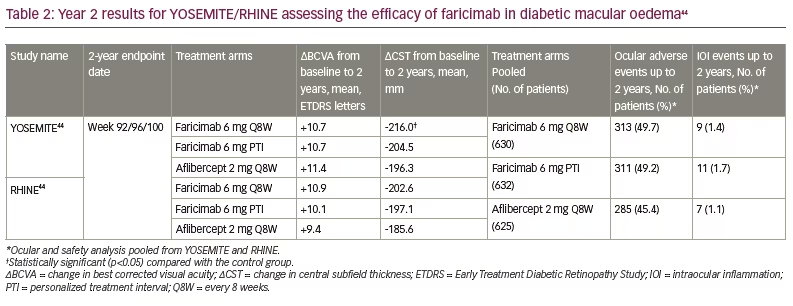
Year 2 data for both YOSEMITE and RHINE were revealed earlier this year, and showed that faricimab maintained non-inferior BCVA gains from baseline compared with aflibercept (Table 2).44 The durability of faricimab was also further supported when examining the PTI cohort, with 78% of patients achieving ≥Q12W dosing and 62% of patients achieving Q16W dosing by Year 2.18 Regarding anatomical outcomes, greater CST reductions from baseline were seen with the faricimab arms through Year 2 compared with the aflibercept arms. In addition, in both studies, patients treated with faricimab achieved earlier resolution of DMO (defined as CST<325 μm) and intraretinal fluid than those treated with aflibercept. The mean number of injections to the first instance of DMO resolution was lowest in the PTI arm (YOSEMITE/RHINE=4.2), followed by the faricimab Q8W arm (YOSEMITE=5.8; RHINE=4.8) and the aflibercept arm (YOSEMITE=7.5; RHINE=5.8).44 Lastly, there were no safety signals noted with faricimab, with low rates of intraocular inflammation and no cases of retinal vasculitis across treatment arms.18,44 Thus, the Year 2 data from YOSEMITE and RHINE support how faricimab is non-inferior from a visual, anatomical and safety standpoint to standard anti-VEGF therapy.
Rhone-X
An extension study for YOSEMITE and RHINE was initiated in 2020 to further assess the long-term safety of faricimab in DMO.43 Rhone-X is a multicentre, 2-year extension study examining the long-term safety and tolerability of faricimab 6 mg in patients who completed the YOSEMITE or RHINE clinical trials. This study is in the data-collection phase and is expected to conclude in August 2023.43
Faricimab in retinal vein occlusion
COMINO/BALATON
With data supporting the use of faricimab in nAMD and DMO, two phase III studies were launched in 2021 to assess faricimab in RVO.19,20 COMINO and BALATON are multicentre, randomized, double-masked comparator-controlled studies assessing the efficacy and safety of faricimab in macular oedema secondary to central RVO/hemi-RVO (COMINO), or secondary to branch RVO (BALATON). The two treatment arms are faricimab 6 mg Q4W or aflibercept 2 mg Q4W until Week 20, with both arms switching to faricimab 6 mg PTI until Week 72. The primary endpoints are change in BCVA from baseline to Week 24 in treatment-naive patients. Both studies have finished recruiting and are collecting data. The estimated primary completion dates for COMINO and BALATON are July and August 2022, respectively.19,20
Conclusion
While anti-VEGF injections have transformed the treatment landscape for retinal diseases like nAMD, DMO and RVO, there are still shortcomings to this approach due to frequent treatment monitoring and dosing. Especially for patients with poor or refractory response to anti-VEGF injections, novel treatments are needed to optimize outcomes in retinal diseases.14,15 With recent FDA approval of faricimab 6 mg for nAMD and DMO,18 as well as clinical trials underway for its use in RVO,19,20 this bispecific molecule is a promising contender for addressing the limitations of anti-VEGF monotherapy. With its dual targeting of VEGF-A and the Ang/Tie-2 pathway that both promote vascular pathogenesis, faricimab has comparable efficacy to mainstay anti-VEGF therapies, with similar safety profiles.39,42 In addition, the enhanced durability of faricimab allows for less-frequent dosing while still maintaining visual gains, thereby reducing treatment burden for patients and providers.39,42
Another novel aspect from the clinical trials is use of a PTI-dosing arm, as seen in YOSEMITE and RHINE.42 The PTI regimen is modelled on the treat-and-extend concept, allowing individualized dosing intervals ranging from Q4W to Q16W according to disease-activity assessments.45 Flexible dosing regimens like PTI were not used in prior landmark trials that led to FDA approval of ranibizumab (ANCHOR, RISE/RIDE)5,46 and aflibercept (VIEW1/VIEW2, VIVID/VISTA)47,48; these followed fixed monthly or bi-monthly treatment dosing.
The main advantage of PTI dosing is that it reflects real-world practice, as providers often treat on a pro re nata (PRN) or treat-and-extend basis according to each patient’s response instead of a fixed schedule.45 This is demonstrated by recent studies that found that patients in real-world practice are often undertreated compared with those in clinical trials.14,15 The potential limitation of PTI dosing is that it may lead to larger retinal CST and fluid fluctuations, which has been associated with poorer visual prognoses.49–52 Thus, while YOSEMITE and RHINE showed that faricimab Q8W and PTI led to comparable visual and anatomical responses, evaluating real-world patient outcomes in DMO and other retinal diseases is warranted in the near future.42
While FDA approval of faricimab was made possible through results from large phase III clinical trials, it is important to note that most trials are only up to Year 1 or Year 2.39,42 While there are extension studies underway, such as AVONELLE-X and Rhone-X, which will assess the effects of faricimab up to Year 4, the long-term efficacy and safety of faricimab are still unknown.40,43 As this is the first FDA-approved molecule with bispecific targeting of both Ang-2 and VEGF-A, the long-term ocular or systemic effects of suppressing the Ang-2 axis are unknown. Notably, a pharmacokinetics study found that faricimab 6 mg led to significantly higher serum concentrations than the approved doses of either ranibizumab 0.5 mg or aflibercept 2 mg, and lower than bevacizumab 1.25 mg.36 In addition, due to faricimab only recently being approved, there are minimal data regarding its potential off-label indications, dosage modifications necessary for patients with other comorbidities, or its efficacy in real-world clinical settings.18
Nonetheless, the clinical trial data thus far support faricimab in successfully targeting multiple pathogenic pathways while maintaining a comparable safety profile to mainstay anti-VEGF agents.39,42 This drug and others like it in development have the potential to revolutionize the treatment experience for patients with retinal diseases.