




The development of anti-vascular endothelial growth factor (VEGF) agents as intravitreal treatments for neovascular age-related macular degeneration (nAMD) has drastically improved the vision outcomes for this disorder. Ranibizumab, a recombinant, humanized monoclonal antibody that targets VEGF-A, was approved in 2006 by the US Food and Drug Administration (FDA) for the treatment of nAMD. The pivotal MARINA1 and ANCHOR2 studies demonstrated the visual efficacy and safety of treatment with monthly intravitreal ranibizumab. Subsequently, the CATT3 and HARBOR4,5 studies showed that monthly ranibizumab dosing was similar to dosing as needed
(pro re nata [PRN]), offering more flexibility for patients. However, despite the option of different dosing regimens, real-world studies have shown that the schedule of frequent and prolonged intravitreal injections required for efficacy is difficult to sustain and burdensome for patients and families.6 In an effort to reduce therapeutic demands, the port delivery system (PDS) was developed to allow the sustained and prolonged release of a special concentrated formulation of ranibizumab. In this review article, we outline the history behind the major studies that affected ranibizumab labelling, and discuss recently published and current studies that have resulted in a new FDA label for ranibizumab.
MARINA and ANCHOR
In the MARINA trial (NCT00056836),1 716 treatment-naïve eyes with minimally classic or occult choroidal neovascularization (CNV) associated with AMD were randomized 1:1:1 to sham (n=238), ranibizumab 0.3 mg monthly (n=238) or ranibizumab 0.5 mg monthly (n=240). At 12 months, eyes treated with ranibizumab 0.5 mg gained 7.2 Early Treatment Diabetic Retinopathy Study (ETDRS) letters compared with baseline and those treated with ranibizumab 0.3 mg gained 6.5 letters, whereas the sham group showed a decrease of 10.4 letters. Given this positive result, ranibizumab was offered to all patients in year 2 and visual acuity benefits were maintained.1 In the ANCHOR trial (NCT00061594),2 423 nAMD eyes with predominantly classic CNV were randomized 1:1:1 to photodynamic therapy (PDT) with verteporfin plus monthly sham injection (n=143), sham PDT plus monthly ranibizumab 0.3 mg (n=140) or sham PDT plus monthly ranibizumab 0.5 mg (n=140). At 12 months, eyes treated with ranibizumab 0.5 mg gained 11.3 ETDRS letters compared with baseline and those treated with ranibizumab 0.3 mg gained 8.5 EDTRS letters, whereas the PDT-treated eyes decreased by 9.5 letters. In both trials, the incidence of endophthalmitis was low, with a reported rate of 0.05% per injection.1,2
These pivotal studies demonstrated the efficacy and safety of fixed monthly intravitreal injections of ranibizumab for nAMD, leading to FDA approval of the 0.5 mg dose of ranibizumab. For the first time, a treatment was available that not only prevented vision loss but led to a mean improvement in vision.
CATT and HARBOR
While the vision improvements demonstrated in the MARINA and ANCHOR trials for what was once a blinding disease cannot be underestimated, monthly, prolonged dosing poses a treatment burden for elderly patients with AMD and their caregivers.6 In 2011, the randomized Comparison of age-related macular degeneration treatment trials (CATT)3 showed that ranibizumab given as needed was equivalent to ranibizumab given monthly with a mean difference of 1.7 letters at 1 year. This was followed by the HARBOR (NCT00891735) trial,4,5 which aimed to investigate whether alternative treatment doses or individualized treatment regimens added further efficacy and/or durability.
In HARBOR, treatment-naïve eyes with nAMD were randomized 1:1:1:1 to ranibizumab 0.5 mg monthly (n=276), ranibizumab 0.5 mg PRN following three monthly loading doses (n=275), ranibizumab 2.0 mg monthly (n=274) or ranibizumab 2.0 mg PRN following three monthly loading doses (n=273). After the three loading doses, the PRN groups were evaluated every month and treatment was given only if specific criteria were met (≥5-letter decrease from the previous visit or any evidence of disease activity on optical coherence tomography [OCT]). At month 24, the mean gain from baseline in best corrected visual acuity (BCVA) was 9.1 letters in the ranibizumab 0.5 mg monthly arm, 7.9 letters in the ranibizumab 0.5 mg PRN arm, 8.0 letters in the ranibizumab 2.0 mg monthly arm, and 7.6 letters in the ranibizumab 2.0 mg PRN arm. The average injection frequency over 2 years for patients receiving ranibizumab PRN was 9.9 weeks, with a mean letter gain of 7.9, and 93% of these patients did not require monthly injections. The study concluded that, at 24 months, the BCVA gains were clinically meaningful and similar among all four treatment arms. These results led to a new FDA label for ranibizumab: “although not as effective, patients may be treated with 3 monthly doses followed by less-frequent dosing with regular assessment.”
Real-life data
The HARBOR study suggested that the response to ranibizumab can be highly individual, as a notable proportion of patients did not need monthly dosing to maintain vision. However, patients in HARBOR were evaluated monthly regardless of study arm, and treatment decisions in the PRN arm were made based on careful ETDRS vision and OCT assessments. In clinical practice, retina physicians often use treat-and-extend administration to balance treatment burden with efficacy and predictability for the patient.6,7 Of note, patients treated with anti-VEGF agents in clinical trials experience greater vision improvements than patients included in observational studies of real-world practice, where visual gains are often limited to less than one line of vision.8–13 Furthermore, data suggest that patients treated in real-world clinical practice are monitored and treated less often than those treated within the setting of clinical trials, which may lead to undertreatment of nAMD and suboptimal vision outcomes.1,2,5,8–14
Paradigm shift with continuous medication delivery
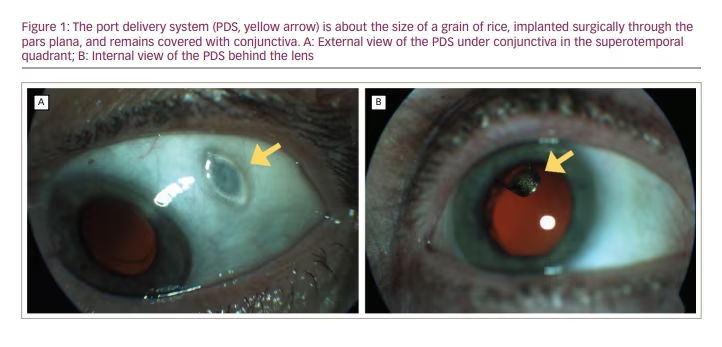
The ranibizumab PDS was designed in an effort to achieve optimal drug delivery with minimal treatment burden. The PDS is a permanent ocular implant that delivers a continuous dose of ranibizumab into the vitreous cavity. The device is approximately 8 mm in length and 2.5 mm in diameter, and is surgically implanted through a scleral incision into the pars plana and covered by Tenon’s capsule and conjunctiva (Figure 1). The procedure requires certain technical modifications of established retinal surgical techniques such as external photocoagulation of the choroid, careful attention to haemostasis, and delicate handling of the conjunctiva and Tenon’s capsule with non-toothed forceps. A highly concentrated investigational formulation of ranibizumab, initially filled at implantation, is then continuously released via passive diffusion from the implant reservoir through a microporous titanium release control element into the vitreous. This results in a controlled continuous release of ranibizumab into the vitreous over time. Refill-exchanges are completed with a proprietary, dual-bore refill wire through a self-sealing septum in the centre of the implant flange. This procedure is facilitated by loupe magnification and can be reasonably accomplished in the clinic.15
LADDER
The LADDER study (NCT02510794)15,16 was a phase II trial that showed initial visual and anatomic success with the ranibizumab PDS over 9 months. The trial included 220 patients randomized in a 3:3:3:2 ratio to receive the PDS filled with ranibizumab 10 mg/mL (n=58), PDS with ranibizumab 40 mg/mL (n=62), PDS with ranibizumab 100 mg/mL (n=59) or monthly intravitreal injections of standard ranibizumab 0.5 mg (n=41), respectively. The mean time to first implant refill was 8.7 months, 13.0 months and 15.0 months in the PDS 10 mg/mL, PDS 40 mg/mL and PDS 100 mg/mL arms, respectively. In the PDS 100 mg/mL arm, 80% of patients went 6 months or more without meeting the criteria for PDS refill. At 9 months, the change in BCVA from baseline was a 3.2 and 0.5 letter loss for the PDS 10 mg/mL and PDS 40 mg/mL arms, respectively. However, for the PDS 100 mg/mL and monthly ranibizumab 0.5 mg groups, there was a 5.1 and 3.9 letter gain, respectively. Mean central foveal thickness change from baseline was similar between PDS 100 mg/mL and monthly ranibizumab 0.5 mg arms at 9 months. The primary adverse event was vitreous haemorrhage, which occurred at a rate of 4.5%. Other adverse events included three cases (1.8%) of endophthalmitis and four cases (2.2%) of retinal detachment in PDS-treated patients.15 Based on LADDER results, the PDS 100 mg/mL emerged as a potential comparable treatment option to monthly intravitreal injection with ranibizumab 0.5 mg at 9 months and led to the phase III ARCHWAY study.17 The study was the first of its kind to demonstrate that biologics can be safely delivered to the eye through a permanent, refillable intraocular reservoir.
ARCHWAY
The phase III ARCHWAY study (NCT03677934)17 was a non-inferiority study with 3:2 randomization comparing the PDS using ranibizumab 100 mg/mL exchanged every 24 weeks (n=248) with monthly intravitreal injections of ranibizumab 0.5 mg (n=167) for nAMD. The PDS arm had a fixed-scheduled drug refill at month 6 with monthly monitoring. If disease activity became apparent in the PDS arm during the study, supplemental injections of ranibizumab 0.5 mg were administered. The primary outcome was change in BCVA from baseline to the average of BCVA from week 36 and week 40. Secondary outcomes were safety, overall BCVA change from baseline and central retinal thickness (CRT) change from baseline.
Results of ARCHWAY were first presented at the 2020 Annual Meeting of the American Society of Retina Specialists.18 The baseline BCVA was 20/32 in both groups following a mean of 5 anti-VEGF injections before baseline. The study had a 98% patient retention rate at the primary analysis. There was a transient reduction in BCVA in the PDS arm that was related to the implantation surgery, with BCVA returning to baseline by week 12. Only 2% of the PDS group required supplemental standard intravitreal injections. At end of study, the PDS 100 mg/mL arm showed a BCVA gain of 0.2 ETDRS letters compared with a gain of 0.5 ETDRS letters in the monthly injection arm. The study concluded that PDS 100 mg/mL with a 6-month refill was non-inferior to monthly ranibizumab 0.5 mg dosing at the primary endpoint average of weeks 36 and 40, as well as at the average of weeks 44 and 48. More than 90% of PDS-treated patients did not receive supplemental treatment before each refill-exchange procedure. In the PDS arm, the most frequent adverse event was a conjunctival bleb (6.5%), vitreous haemorrhage with spontaneous resolution (5.2%) and conjunctival retraction and erosion (4.4%). Four cases (1.4%) of endophthalmitis related to conjunctival retraction developed. The vision loss was irreversible in one case but returned to baseline for the remaining three cases. Two retinal detachments requiring surgery occurred. There was also one case of device dislocation into the vitreous cavity during a refill-exchange procedure; the device was removed, and the vision returned to baseline. No traumatic cataracts were noted.
Overall, the PDS with ranibizumab 100 mg/mL was well tolerated and represents a novel drug-delivery modality comparable to monthly ranibizumab 0.5 mg for patients with nAMD. On 22 October 2021, the FDA approved the PDS for patients who have previously responded to at least two intravitreal injections of an anti-VEGF medication.
PORTAL
The PORTAL study (NCT03683251)19 is an ongoing extension phase III study looking at the long-term safety profile in patients receiving PDS 100 mg/mL from the LADDER and ARCHWAY trials. The study plans to evaluate patients with PDS receiving clinic-based refills every 24 weeks for 144 weeks, with bimonthly monitoring visits. Results are anticipated in several years following the rollover of all the phase III patients into this extension trial, and there will be ongoing reporting of efficacy and safety data. This study will be crucial in monitoring long-term potential implant benefit, as well as related safety issues, such as endophthalmitis, conjunctival breakdown with PDS exposure, implant dislocation and retinal detachment.
Conclusion
Intravitreal anti-VEGF injections have been used in the clinical setting since 2004 with the approval of pegaptanib sodium, and ranibizumab has been in use since 2006. These treatments are part of the anti-VEGF toolkit that has saved the vision of millions of patients with nAMD. The PDS is the first long-acting drug-delivery system with complete and promising phase III data. Although patients are still currently required to attend the clinic for monitoring visits, which were set at monthly for the ARCHWAY study and bimonthly for the PORTAL study, the concept of continuous medication delivery could lead to the first paradigm shift in the treatment of AMD since the inception of intravitreal anti-VEGF injections. However, the accumulation of long-term safety and tolerability data from studies such as the PORTAL trial and subsequent real-world observational data will be crucial in evaluating the adoptability of this new technology into routine clinical practice.


