Proceedings of the inaugural European LHON Forum Meeting, held in Frankfurt, Germany
Leber’s hereditary optic neuropathy – the biology behind this deceptively complex disease
In the first plenary session co-chaired by Prof Valerio Carelli and Dr Catherine Vignal-Clermont, the pathophysiology, epidemiology and symptoms typically experienced by patients with Leber’s hereditary optic neuropathy (LHON) were discussed.
LHON was first described by Albrecht von Graefe in 1856, and defined as a clinical entity in 1871 by Theodore Leber.1,2 The pathophysiology of LHON involves selective dysfunction of retinal ganglion cells (RGCs), with preferential degeneration of the smallest calibre RGC fibres,3 and later apoptosis of these cells due to reduced adenosine triphosphate (ATP) production and increased free radical production, possibly in a synergistic pattern, as both factors are believed to be intrinsically related.4,5,6,7
LHON is the most common inherited mitochondrial (mt) DNA disorder. Over 90% of LHON are a result of pathogenic point mutations at mtDNA positions G11778A, G3460A or T14484C, which code for subunits of the mitochondrial respiratory chain complex I (Figure 1).8,9 In adult-onset disease, the T14484C mutation is associated with a more benign disease course with a substantially higher frequency in spontaneous recovery of vision than other mutations.10 The G11778A and G3460A mutations are associated with a more severe clinical outcome.10 There is a ~40–70% chance of some degree of visual improvement with the T14484C mutation.11 However, visual improvement is less frequent with the G11778A and G3460A mutations (4–20%).11,12 By comparison, in childhood-onset LHON, spontaneous visual recovery is thought to be more common in patients carrying the G3460A or T14484C versus the G11778A mutation.13
Figure 1: Membrane complexes in the mitochondrial respiratory chain8,9
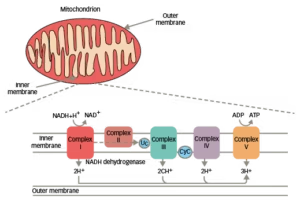
ADP = adenosine diphosphate; ATP = adenosine triphosphate; CyC = cytochrome C; LHON = Leber’s hereditary optic neuropathy; NAD+/NADH = nicotinamide adenine dinucleotide.
Other rare pathogenic mtDNA point mutations, as well as unusual clustering of mtDNA polymorphic variants were reported to account for the remaining 10% of patients with LHON not carrying one of the three common mutations.14,15
Furthermore, other mtDNA variants, referred in the past as ‘secondary’ mutations, now classified as diagnostic markers of mtDNA haplogroups, are also implicated as modifiers of LHON penetrance within the maternal lineages.3,9,11,16
In addition to the recognised role of mtDNA mutations and haplogroups, additional nuclear genetic factors, and/or hormonal and environmental factors may impinge on disease expression.11,17 There is convincing evidence that heavy smoking,18,19 and to a lesser extent excessive alcohol consumption,19 are risk factors for visual loss in LHON. Whether the gender bias is due to hormonal differences, especially the higher levels of circulating oestrogen in women, has to be further confirmed as there is limited evidence of a second peak of female LHON carriers converting in the peri-menopausal or menopausal period.17
Prevalence of LHON is not well known but it is estimated at between 1/15,000–1/50,000 people worldwide.20 The overall male to female ratio among patients with LHON averages around 4:1, but this can vary depending on the primary mutation.21,22 The lifetime risk of a male mutation carrier losing vision is ~50% compared with ~10% for a female carrier.17
Adult-onset LHON (15–35 years of age) usually presents as painless, subacute central visual loss in one eye, followed by involvement of the second eye within 1 year in 97% of those affected.23,24 In most patients, vision loss is devastating in terms of the impact on their quality of life, and remains permanent.11,25 Childhood onset (≤10 years) is less often reported, and is generally associated with a better prognosis than adult-onset LHON for all mutations.13,23 Overall, approximately one in five patients will remain within the visual acuity (VA) criteria for legal blindness.13
In rare cases, extraocular features of LHON can present, such as peripheral neuropathy, ataxia, tremor, myoclonus, cardiac conduction defects and myopathy.26
The patient’s long and winding road to diagnosis – streamlining the diagnosis of LHON
In the second plenary session, co-chaired by Prof Thomas Klopstock and Dr Patrick Yu-Wai-Man, the challenges in diagnosis of LHON were addressed and a patient with LHON shared his diagnostic journey with the attendees.
Differential diagnosis
Diagnosis of LHON is often delayed, because of the relative rarity of the disease and the heterogeneous presentation.3,17 LHON disease progression can be characterised by four different stages (Figure 2).11,17,27,28
Figure 2: Phases of LHON11,17,27,28
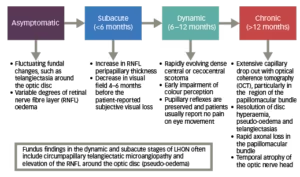
The clinical stages of LHON can also be defined according to time from onset and clinical investigations (VA, visual fields, optical coherence tomography [OCT]).28 If a patient is only seen at the chronic stage, diagnosis can be challenging if there is no clear maternal family history.27 As a result, other compressive, infiltrative and inflammatory causes of a bilateral optic neuropathy can sometimes be mistakenly considered as a potential cause.27
In the less common childhood-onset LHON, the majority of patients display the classical dynamic pattern of visual loss and progression (~60%), with the remainder either following a slowly progressive or insidious/subclinical pathway.13 Diagnostic delays of up to 15 years have been reported in children with an insidious mode of onset.13
Diagnosis of LHON can usually be made based on patient and family clinical history as well as baseline investigations including a formal neuro-ophthalmological examination and mtDNA genetic testing (Figure 3).28,29 Because the three primary LHON mutations account for the vast majority of cases of LHON, they should be the first to be screened in a patient with vision loss suspicious for LHON.11 If none of these three mutations are detected, and clinical suspicion remains high, then sequencing of the entire mtDNA to disclose the rare mtDNA mutations is indicated, particularly when a family history of LHON-like disease is elicited.11
Figure 3: Differential diagnostic flowchart for LHON28,29
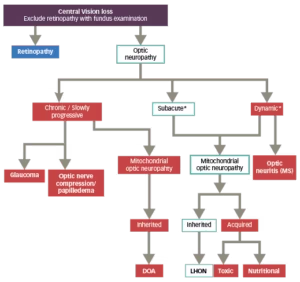
Adapted from Sadun et al., 2012 and Carelli et al., 2017.
*The evolution of LHON in the first weeks/months is described canonically as dynamic/subacute, depending on how rapidly the loss of visual acuity evolves.<sup”>2
DOA = dominant optic atrophy; LHON = Leber’s hereditary optic neuropathy;
MS = multiple sclerosis.
Other optic neuropathies, especially optic neuritis (neuromyelitis optica), toxic, metabolic and compressive optic neuropathies, maculopathies and nonorganic visual loss (not a diagnosis of exclusion) should be excluded.28
OCT has enabled objective confirmation and quantification of the characteristic fundus changes in LHON.11 OCT assessment of the retinal nerve fibre layer (RNFL) shows thickening during the dynamic phase of the disease and temporal to global thinning during the chronic phase.30
The electrophysiological characteristics of patients with LHON include marked reduction in the N95 component of the pattern electroretinogram (PERG) and markedly abnormal to no detectable P100 component of the pattern visual evoked potentials.31
Decrease of PERG N95 amplitude may already be seen in the initial stages of LHON, whereas in other optic neuropathies, N95 loss is normally seen only after retrograde degeneration of ganglion cells. Improvement of vision may be associated with faster optic nerve conduction reflected in shortening of VEP-P 100 latency, suggesting remyelination.31
Magnetic resonance imaging (MRI) can be used to visualise white matter or other types of lesions, as well optic nerve enhancement with gadolinium (rare in LHON), which can help differentiate between multiple sclerosis (MS), other neurological conditions and LHON.32
In some patients, LHON is associated with multisystem involvement, which may be subclinical depending on age, ethnicity and possibly the heteroplasmy load (the presence of more than one type of mtDNA) of the responsible primary LHON mutation.33 Heteroplasmy, which rarely occurs in LHON families,34 is the result of a purifying germline mechanism known as ‘the mtDNA bottleneck’,3 which leads to a random shift of mtDNA mutational load between generations and is responsible for the variable levels of mutated mtDNA observed in affected offspring from carrier mothers with pathogenic mtDNA LHON mutations.
LHON-multiple sclerosis – Harding’s disease
Central nervous system inflammatory demyelination, compatible with a diagnosis of MS, and LHON have been observed in some patients,35 and is referred to as Harding’s disease. It has been suggested that the co-occurrence of MS and LHON mtDNA mutations is likely to be due to chance.36 However, the resulting disorder (LHON-MS) has a distinct phenotype, implicating a mechanistic interaction.36 LHON-MS differs from classic LHON and typical MS in a number of ways. Although the age at onset and bilateral sequential visual involvement and lack of visual recovery observed in LHON-MS is also typical in LHON, LHON-MS more commonly affects women (2.1:1) and is associated with a longer time interval before both eyes become affected (average ~2 years, and up to 17 years).36,37 When comparing LHON-MS with MS, shared features are age at onset and higher prevalence in women.36 However, LHON-MS is associated with a higher proportion of patients with visual involvement, lack of ocular pain and absence of visual recovery, particularly after therapy with steroids, in most patients.36
Treating LHON – the beginning of a new chapter
The third plenary session, co-chaired by Prof Marko Hawlina and Prof Alfredo Sadun, addressed the latest understandings in the treatment of LHON and examined the roles of pharmacological and gene therapy.
The sudden onset of severe visual loss due to LHON, frequently in young healthy adults, can cause significant distress and anxiety for both patients and their families.3 Male LHON carriers can be reassured that their children are not at risk of inheriting their genetic defect, since the mitochondrial genome is maternally inherited.3 Female carriers will transmit the mutation to all their children, but the level of the mtDNA mutation transmitted can fluctuate if the mutation is heteroplasmic.3
Pharmacological therapy
Patients with LHON are often treated with high-dose steroids before a molecular diagnosis has been established, and this can be used to exclude the possibility of an inflammatory optic neuropathy, as this treatment has no impact on the symptoms of the disease.3 Steroids also do not prevent the subsequent involvement of the fellow unaffected eye.3
Idebenone, a synthetic short-chain benzoquinone that acts as a potent antioxidant and inhibitor of lipid peroxidation,38 is the first and only disease-specific therapy approved for the treatment of visual impairment in patients with LHON.39,40 It facilitates electron flux directly to complex III, bypassing the dysfunctional complex I of the mitochondrial respiratory chain, thereby increasing ATP production.38 As a result, it is thought to protect against the loss of RGCs, as well as the reduction in retinal thickness and gliosis associated with LHON, with the potential to restore the functional loss of vision.41
The treatment objectives with idebenone in patients with LHON are to achieve clinically relevant stabilisation by prevention of further VA loss in patients with good residual VA at baseline or clinically relevant recovery (CRR) of VA in patients at a more advanced disease stage (Figure 4).42
Figure 4: Treatment objectives for Leber’s hereditary optic neuropathy42

Treatment with idebenone has been shown to be well tolerated and associated with demonstrated efficacy.4 In the largest phase III multicentre, double-blind randomised controlled trial in LHON (the Rescue of Hereditary Optic Disease Outpatient Study [RHODOS], n=85), the pre-specified primary endpoint, to achieve a significant improvement in best-corrected visual acuity versus placebo at 24 weeks in all patients enrolled was not achieved, but all of the secondary endpoints showed a positive trend towards visual improvement.43 A responder analysis, however, showed rates of CRR at week 24 were also numerically higher with idebenone than with placebo (30.2% versus 10.3% of patients). Patients who previously could read some letters on the chart were assessed as having CRR when they could read ≥10 more letters (two lines), and those who previously could not read any letters (i.e. ‘offchart’) were assessed as having CRR when they could read ≥5 letters (one line) on the chart.38 It should be noted that the majority of patients enrolled had disease onset of >12 months (n=58), where a therapeutic effect would be more challenging. In an open-label follow up of the RHODOS trial, a beneficial effect of 6 months treatment with idebenone persisted for a median time of 30 months after the discontinuation of therapy.44 Further supportive evidence on the long-term effects of idebenone in people with LHON was presented. In a retrospective analysis of patients participating in an Expanded Access Program (EAP) (manuscript submitted) the proportion of people achieving a CRR from nadir reached 47.1% and the mean time to initial observation of a CRR was 10.0 months (range 2.5–26.5 months). In some patients an initial deterioration of VA was observed (and reached a nadir) after treatment, which may still result in a CRR after treatment maintenance. Another interesting observation was that the magnitude of the VA in those patients that show CRR can further improve with treatment maintenance, and achieving an average of up to seven lines in the Early Treatment Diabetic Retinopathy Study (ETDRS) chart after a mean of 28.7 (3.0–58.2) months.
Regarding maintenance of residual vision in patients with VA not yet in the threshold of legal blindness, defined as clinically relevant stabilisation (patient having a logMAR of <1.0 at baseline and maintaining a logMAR of <1.0 at the last follow-up assessment), results showed that of the 87 people in the EAP, 24 had a VA at baseline <1.0 logMAR, and 50.0% (12/24) maintained this at last observation after a mean (±standard deviation [SD]) treatment duration of 28.2 ± 17.2 months (range 9.9–58.0 months). In addition, a retrospective Italian study (n=103) confirmed that patients receiving idebenone were more likely to recover vision if treatment was initiated early and if it was maintained for longer than 24 weeks.12
The expert consensus recently published is that idebenone should be started as soon as possible at 900 mg/day in patients with disease less than 1 year in duration.28 There is not enough evidence to recommend treatment in patients with chronic disease 1–5 years after the second eye onset, and no evidence to recommend treatment in patients with chronic LHON present for >5 years after the second eye onset.28 Treatment with idebenone should continue for at least 12 months before evaluating if there is response to therapy. If there is no clinically relevant response after 1 year, interruption should be considered. However, the data from the EAP indicate that first response to treatment may take up to 30 months. Once efficacy has been confirmed, treatment must be continued until a plateau is observed and from that point, treatment maintained for another year before considering discontinuing it.
Gene therapy
LHON could be amenable to gene therapy. However, mitochondrial gene therapy is challenging because of the physical barrier imposed by having both inner and outer mitochondrial membranes, and the need for sustained gene expression following delivery of the gene construct into the mitochondrial matrix compartment.26 Genetically modified adeno-associated viral vectors (AAV2) have been developed to deliver a gene construct as a therapeutic strategy to compensate for the G11778A mutation, and preclinical data suggest a resulting therapeutic rescue in vitro and a reduction in the amount of RGCs lost in the murine models tested.45
Two separate randomised, double-masked, sham-controlled pivotal phase III trials designed to evaluate the efficacy of a single intravitreal injection of GS010 (AAV2 that codes for the wild-type ND4 protein) in patients affected by LHON due to the G11778A mutation in the mitochondrial ND4 gene are being conducted (REVERSE [NCT02652780] and RESCUE [NCT02652767]). The primary endpoint is the difference in changes from baseline on the ETDRS chart at 48 weeks post-injection. Early results from REVERSE have been inconclusive, with no differences between treated and sham eye.46 Results from the RESCUE trial are expected in 2019. Another trial, the multicentre, randomised, double-masked, placebo-controlled study REFLECT (NCT03293524), is currently recruiting patients.
The burning question is whether gene therapy will be effective and if so, at what phase (pre-clinical or clinical) of the disease should it be utilised. Another question is whether it should be combined with idebenone, in order to treat the disease from different pathophysiologic approaches. At the current time, there is no cure for LHON.20 However, finding a cure is still the primary objective of current research. In order to achieve this, greater numbers of patients are needed for clinical trials, and recently established patient registries are aiding this.
Beyond LHON – a wider view of optic neuropathies
The final plenary session, co-chaired by Prof Valerio Carelli and Dr Catherine Vignal-Clermont, discussed the challenges of differentiating LHON from other optic neuropathies and occult retinopathies and reviewed the progress in our understanding of the genetic aspects and latest research into the pathophysiology of LHON.
Mitochondrial optic neuropathy can occur in isolation, as in the case of LHON, but can also be a part of a more general neurological syndrome or acquired as a result of environmental stressors. Considerable progress has been made in the understanding of optic neuropathies, which can present as unilateral or bilateral vision loss, simultaneously or sequentially.3,17 Despite this, differentiating LHON from other optic neuropathies can be challenging and requires careful exclusion of toxic entities and optic neuritis.
LHON involves subacute and painless visual loss with central scotoma and poor colour vision with sequential involvement of both eyes over a period of weeks to months.47 Funduscopic examination mainly shows circumpapillary telangiectasia.47 Optic neuritis is typically associated with fat-supressed orbital MRI enhancement, unlike LHON, which is notable for often normal MRI images, but which may reveal non-specific white matter lesions.32,47Exposure to toxins closely associated with optic neuropathy should be investigated, including carbon monoxide, ethylene glycol, perchloroethylene, methanol and tobacco.47
If the ophthalmologic assessment, including an assessment of VA, colour vision, visual fields and electrophysiology, as well as molecular genetic testing leave any uncertainty about the diagnosis of LHON, further investigations are appropriate to exclude other potentially reversible causes of bilateral optic neuropathy.48 Depending on the clinical presentation and evolution, autoantibody testing and an infectious or vasculitic screen may be warranted.48
In recent years, many mtDNA mutations that are associated with several clinical manifestations across genetics, oncology, neurology, immunology and critical care medicine have been identified.49 The availability of therapy for LHON has boosted interest in the research of other mitochondrial diseases. However, due to the rarity of LHON, it has been difficult to study and there remain many unanswered questions regarding its pathophysiology. One area that is being actively researched is tissue specificity. Typically, only the optic nerve is involved in LHON, with a specific pattern of optic atrophy due to the primary loss of the papillomacular bundle (PMB), which is the most vulnerable region in LHON especially at the inferior temporal region of the optic disc, where the unmyelinated axons are the smallest.22 The key to this preference for the PMB appears to be in the energetics imparted by axon calibre and myelin.22 Myelin allows for faster conduction and provides a more energy-efficient method for re-establishing membrane potential after an action potential, hence myelinated axons use less ATP and produce less reactive oxygen species.22 In the investigation of the impact of metabolic stress on an axon in relation to its size, a mathematic model confirmed that smaller axons were more susceptible to stress and therefore preferentially destroyed.50 This research suggests that RGCs with smaller axons would be expected to have a reduced ability to transport mitochondria further along the axon, and this means that areas requiring higher mitochondria concentrations, such as the RNFL, laminar and prelaminar regions of the optic nerve head, the nodes of Ranvier in the post-laminar portion and synaptic terminals, could be more susceptible to metabolic and functional defects.22